材料与仪器
cDNA 模板、AllTaq Master Mix Kit;
HindIII 和 XhoI 酶和 buffer;
QIAquick Gel Extraction Kit;
无缝克隆同源重组试剂盒;
QIAprep Spin Miniprep Kit;
大肠杆菌感受态细胞 DH5α;
摇床、离心机、PCR 仪等设备。
步骤
1. 从 cDNA 模板中扩增目的基因并添加同源重组臂
设计带有同源重组臂的序列进行 PCR 扩增:
上游引物:TAGCGTTTAAACTTAAGCTT+ATGXXXX(前 20 个碱基为载体上的同源重组臂,ATG 是起始密码子)
下游引物:ACGGGCCCTCTAGACTCGAG+TTAXXX(前 20 个碱基为载体上的同源重组臂,TTA 是终止密码子对应的序列)
PCR 反应体系:
|
组分 |
用量(50ul 体系) |
|
AllTaq Master Mix,4× |
12.5ul |
|
上游引物(浓度 10uM) |
1.2ul |
|
下游引物(浓度 10uM) |
1.2ul |
|
模板 cDNA |
0.1pg-1ug |
|
无酶水 |
补齐至 50ul |
PCR 程序:
|
步骤 |
温度 |
时间 |
|
预变性 |
95℃ |
2 min |
|
变性 |
95℃ |
5s |
|
退火 |
55℃(根据需要可调整) |
15s |
|
延伸 |
72℃ |
10s |
后三个步骤循环 35-40 次
注:如果引物扩增效率偏低,可以先根据目的基因的序列设计特异性引物,从 cDNA 中扩增目的基因片段,然后再以回收片段为模板,用带有同源臂序列的引物进行扩增,获得的片段用于构建载体。
2.pcDNA3.1+载体使用 HindIII 和 XhoI 进行双酶切
|
组分 |
用量 |
|
质粒载体 |
1ug |
|
HindIII 酶 |
1ul |
|
XhoI 酶 |
1ul |
|
Buffer |
两种酶兼容缓冲液(如 CutSmart Buffer 10×,添加 5ul) |
|
无核酸酶水 |
补齐至 50ul |
37℃ 酶切 1 h
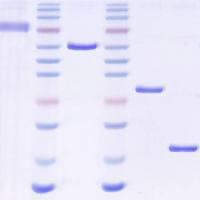
3. 琼脂糖凝胶电泳分离 PCR 扩增的目的片段及线性化载体
配置新鲜的 1×TAE 电泳缓冲液,配置 1% 浓度的琼脂糖凝胶用于电泳,电泳强度不超过 5V/cm,电泳时间根据条带迁移的情况而定,一般 30 分钟左右。
4. 胶回收纯化线性化质粒及 PCR 扩增片段
切胶回收目标 PCR 产物条带和线性化质粒条带,回收流程参考如下:
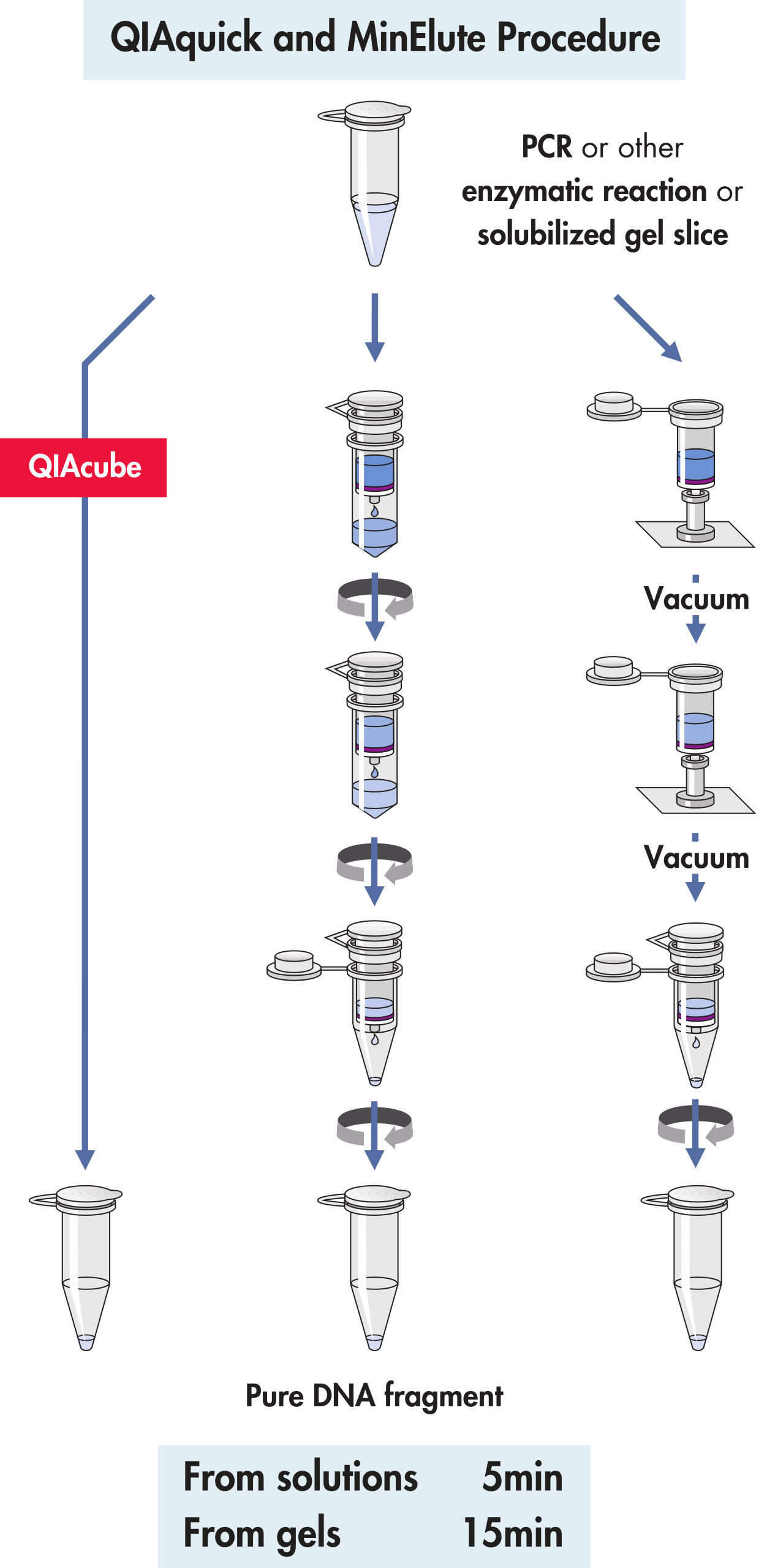
|
切胶称重加入 buffer QG |
BufferQG 按照胶 3 倍体积添加(100 mg 胶≈100ul) |
|
溶胶 |
50℃ 温浴 10 min,间隔摇晃混匀,如果颜色变紫色,可以添加 10ul 3M 的醋酸钠调节 pH 至颜色变为黄色 |
|
加异丙醇 |
加入一倍胶体积的异丙醇,混匀 |
|
纯化柱纯化 |
溶液加入 QIAquick 纯化柱,离心,弃滤液或真空吸滤液 |
|
漂洗 |
加入 750ul Buffer PE 至纯化柱,离心弃滤液 |
|
洗脱 |
加入 50ul buffer EB 或者无核酸酶水,离心收集滤液 |
回收 PCR 产物和线性化质粒进行 Nanodrop 或取微量进行琼脂糖凝胶电泳来检测回收 DNA 的质量。
5. 目的片段及线性化质粒进行同源重组
|
组分 |
用量 |
|
线性化载体 |
约 100ng 可根据重组酶说明书建议调整 |
|
目的片段 |
约 80ng 可根据重组酶说明书建议调整 |
|
5×重组 buffer |
4ul |
|
重组酶 |
2ul 或根据重组酶说明书建议调整 |
|
无核酸酶水 |
添加至 20ul |
冰上配置体系,后于 37℃ 孵育 30 min 后冰上冷却备用。
6. 连接产物转化大肠杆菌
|
取大肠杆菌感受态 DH5α |
冰上解冻 |
|
连接产物转化 |
10ul 连接产物冰上加入 100ul 感受态细胞,混匀孵育 30 min |
|
热激 |
42℃ 热激 45s-90s,迅速冰上冷却 3 min |
|
复苏 |
加入 900ul 液体 LB 无抗培养基,37℃,200rpm,1 h |
|
涂板 |
低速离心,弃 800ul 培养基,剩余培养基重悬菌体涂板 |
|
培养 |
37℃ 倒置培养皿培养 12-16 h |
7. 大肠杆菌培养及挑选单克隆菌落
挑选大小正常,表面光滑圆润的单克隆至液体 LB 培养基(含氨苄抗生素)中培养,37℃,200rpm,培养,菌液浑浊后即可取 1ul 菌液用于 PCR 鉴定,其余菌液可继续培养至 12 h。
8. 单克隆菌 PCR 鉴定
参考前文的 PCR 反应体系和程序,检测引物可以使用特异性引物或者载体上的通用测序引物,或者一条特异性引物加一条测序引物。如果单克隆为阳性,可以在琼脂糖凝胶电泳检测时,看到单一明亮的条带。如果条带大小与预期不符,或者无条带,条带过弱,均为假阳性克隆,则丢弃对应的菌液。
9. 质粒小提及测序鉴定
质粒小提可参考如下操作流程:
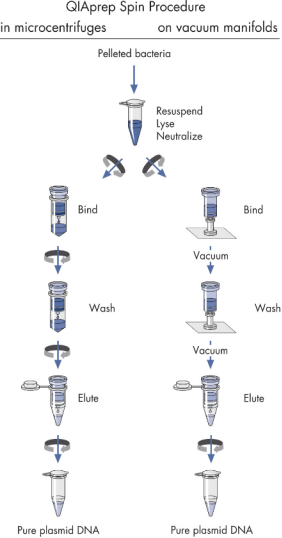
|
菌体收集 |
2 ml 菌液 8000rpm 室温离心 3 min,弃上清 |
|
菌体重悬 |
收集的菌体加 250ul 溶液 P1,重悬混匀 |
|
菌体裂解 |
加入 250ul 溶液 P2,轻柔上下颠倒混匀至溶液澄清 |
|
溶液中和 |
加入 350ul 溶液 N3,轻柔上下颠倒混匀至出现白色沉淀 |
|
离心沉淀 |
室温下 13000rpm 离心 10 min,沉淀积于离心管底部 |
|
过纯化柱 |
去上清液加入纯化柱,13000rpm 离心 30s 弃上清 |
|
去除蛋白 |
加入 0.5 ml 的溶液 PB 漂洗纯化柱,13000rpm 离心 30s 弃上清 |
|
漂洗质粒 |
加入 0.75 ml 的溶液 PE 漂洗纯化柱,13000rpm 离心 30s 弃上清 |
|
去除乙醇 |
13000rpm 离心 60s,去除膜上残留的漂洗液、乙醇等杂质 |
|
洗脱质粒 |
向膜中心加入 50ul 的 buffer EB 或无核酸酶水,孵育 1 min,13000rpm 离心 1 min,洗脱质粒 |
注:
1. 溶液 P1 在使用之前要加入 RNase A,并且加入 RNase A 后需要将其保存于 4℃;
2. 在溶液 P1 中,如果添加了 LyseBlue 颜色指示剂,在 P2 加入后,颜色变蓝色,N3 加入后,颜色变澄清无色;
3. 去蛋白液 Buffer PB 一般在使用的感受态细胞是 endA+基因型时需要加入,用于去除残留较多的核酸酶;
4. 漂洗缓冲液 PE 在使用之前要按照比例添加酒精;
5. 为提高洗脱效率,buffer EB 或者无核酸酶水洗脱质粒之前可以温浴至 65℃ 再加入到硅胶膜中心用于洗脱质粒。
内容来源:QIAGEN 凯杰
来源:丁香实验