


DNA 的定量与连接反应:纯化的gusA 片段与经BamHI/HindIII 作用的pQE31 片段混合后,末端序列互补的部份会进行黏合,但仍须藉由DNA ligase 催化phosphodiester bond 的形成,将缺口接合起来。
利用冰冷的CaCl2 制备competent cells以传统方法进行质体的转形。 仪器用具:37℃培养箱;42℃水浴;冰浴 药品试剂: 0.1 M CaCl2置冰浴中 2 M glucose: 取18 g glucose溶于水中,并调体积至50 mL,无菌过滤,分装后置 -20℃贮存。 2 M Mg2+: 取20.3 g MgCl2 · 6H2O及24.65 g MgSO4 &mi ...
取80 mL SOB培养基装入250 mL灭过菌的三角瓶。 另取1 mL SOB加于微量离心管中。由SOB固体培养基上选8 个约2~3 mm大小的单一菌落接入1mL SOB中,震荡将菌体打散后,加入80 mL SOB中,于37℃震荡培养约2~3h,直至细胞数约为4~7×107/mL.
X-Gluc 可被GUS作用而释出蓝色的产物,若转型株所带的质体接有gus 基因的重组质体,则应可在IPTG 的诱导条件下表现出蛋白质,若此重组蛋白质具有GUS 活性,可将进入菌体的X-Gluc 分解而使菌落呈蓝色
mRNA提取纯化
Precellys组织匀浆器
高通量提取植物组织
组织匀浆器
这个方法是以phenol 及chloroform 溶菌后直接进行电泳分析,并未将质体DNA与宿主细菌染色体DNA 分离,得到的DNA 也不能用限制酶作用。
在胶体中的DNA 可利用毛细现象的作用将DNA 牵引转印至覆盖在胶片上的滤膜,经烘烤或UV 照射后,可使DNA 固定在膜上,以进行探针的杂合(hybridization) 反应。
之前已分别构筑出GUS 基因的表现质体pQG11若要令启动子T5promotor 的启动受到更好的调控,应进一步将pQG11 转形至大肠杆菌M15. M15 可持续表现lac repressorpQG11 上用以驱动GUS基因表现的T5 promoter 的活性将因而受到抑制,但一旦加入IPTG 就能诱导GUS基因大量表现。 在这一节的实验里,我们将分别自经IPTG 诱导与未经诱导的pQG11/M ...
由于RNA聚合酶一般由数个次单元组合而成,早先要在试管内应用RNA聚合酶的转录活性合成RNA并非易事,但这个问题在SP6, T3 及T7 等噬菌体的RNA聚合酶陆续被发现以后,情况已完全改观。
DNA聚合酶要进行DNA聚合反应时一定要有引子 (primer),因为它只能以引子所提供的3'-OH为起始点进行延伸 (extension) 的工作,而不能就地重新合成新的DNA (de novo synthesis)。
以phenol/chloroform 进行DNA 萃取:进行这一部份实验的前,请先确定pBlueGus/HindIII 与pBlueGus/BamHI 两者的酵解反应都已经完全。
文章作者介绍了一种基于脂质体的高效siRNA转染方法。利用该protocol,可以在CCE细胞达到98%的转染效率,在D3细胞达到80%的转染效率,并且对干细胞的形态没有影响。
DNA实验室的故事之一:远古DNA
老外教你跑胶(琼脂糖凝胶电泳)
手机医疗专家
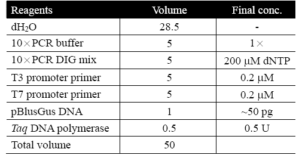
PCR是极为重要的DNA增殖技术,应用层面非常广,其中包括了核酸探针的制备。如果一段DNA已经被次选殖至载体上,要以PCR扩增这段DNA时,可根据质体multiple cloning sites两边的序列,选用适当的核酸引子 (universal primers) 进行扩增反应;比较常用的引子包括SP6, T3 与T7 promoter primer, M13/pUCforward与reverse primers等;若有特殊考量,也可以使用序列专一性核酸引子对。进行PCR反应时若同时添加 [α-32P]dNTP或DIG-11-dUTP,所增殖的DNA即被32P或DIG所标定,所以PCR是制备核酸探针非常便捷好用的方法。
mRNA为单股,一般会卷绕成特定的三级结构,并与某些蛋白质结合,而以messenger ribonucleoprotein particles (mRNPs) 的型式存在于细胞内;自细胞抽取RNA时,一般会以特定步骤去除蛋白质,但仍难完全破除RNA的二/三级结构。 由于进行电泳分析时,影响核酸泳动速率的因子主要包括核酸的长度与构形 (conformation),为了去除核酸构形所造成的影响,一般以电泳分析RNA时都采用变性胶体电泳;在进行这一类电泳分析时,由于RNA已经被变性 (denature),影响其泳动速率的因子主要是长度。 一旦RNA以变性胶体电泳解析好,即可进一步进行北方转印与杂合分析。
关于丁香通
公司信息
个人用户
企业机构


