肿瘤领域研究方向之肿瘤治疗与耐药
百奥智汇
肿瘤治疗与耐药机制研究是肿瘤研究和临床实践中极为关键的领域,具有深刻的生物学意义与临床转化价值。肿瘤治疗的核心目标在于通过手术、化疗、放疗、靶向治疗及免疫治疗等多种手段协同干预,最大程度清除肿瘤细胞,以延长患者的生存期并提升生活质量。然而,耐药性的产生始终是制约治疗效果的关键瓶颈。耐药性可以分为先天耐药(治疗前已存在耐药克隆群)和获得性耐药(治疗过程中适应性演化形成的新耐药机制)两类,其分子机制具有高度异质性:包括驱动基因的新发突变或扩增、表观遗传修饰重塑、代偿性信号通路激活、药物外排泵(如 ABC 转运蛋白家族)表达上调、肿瘤细胞免疫原性降低导致的免疫逃逸,以及肿瘤微环境(如纤维化基质屏障、免疫抑制细胞浸润)的保护性作用等。耐药性的出现不仅直接削弱治疗应答,更可能成为肿瘤复发与远处转移的潜在驱动因素。
因此,系统解析肿瘤耐药的演化轨迹与分子机制,开发基于联合用药的耐药逆转策略,发掘新型治疗靶点及疗效预测生物标志物,对于提升肿瘤治疗的精准度与持久性、改善患者预后具有不可替代的临床意义。
接下来,我们将通过具体案例,阐述如何利用单细胞与空间组学技术解析肿瘤治疗及耐药相关的生物学问题。
案例一:多组学分析揭示肝癌起始细胞免疫逃逸机制和精准治疗策略

发表期刊:Cancer Cell
影响因子:44.5
发表时间:2024 年 12 月
研究疾病:肝细胞癌(Hepatocellular Carcinoma,HCC)
样本类型:患者来源的肝癌类器官(HCC organoids),肝癌细胞系(Hep3B,Huh7,PLC/PRF/5, MHCC97 H,Hep53.4)和小鼠模型(包括自发性肝癌模型和异种移植模型)
样本数量:11 个 HCC 类器官样本,4 个肝癌细胞系样本和小鼠模型(每组至少 6-8 只小鼠)
样本分组:临床样本按 CD155 表达水平分为高表达组与低表达组
应用技术:单细胞 RNA/TCR 测序(scRNA/TCR-seq)、单细胞蛋白组学(CITE-seq)、单细胞染色质可及性测序(scATAC-seq)、bulkRNA 测序、bulkATAC 测序
研究背景
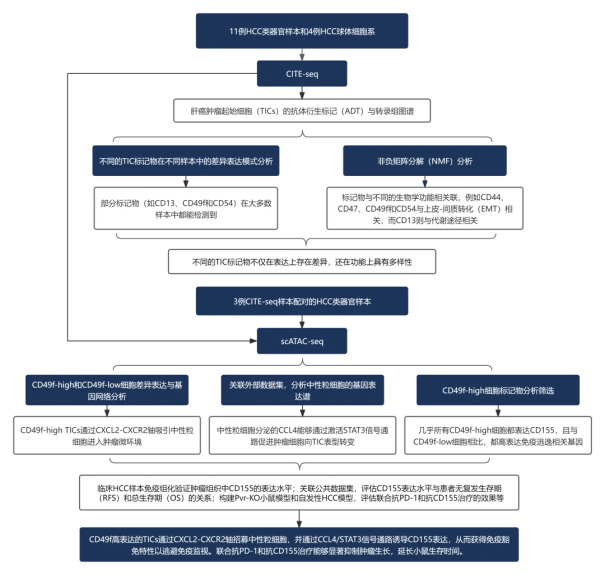
肝细胞癌(HCC)具有显著的细胞异质性,传统治疗疗效有限,免疫治疗在 HCC 中也面临应答率低等挑战。肿瘤起始细胞(Tumor-Initiating Cells,TICs)是一类具备干细胞特性和高致瘤潜力的细胞亚群,可通过逃避免疫监视介导免疫治疗耐药,被认为是 HCC 免疫治疗失败的关键原因之一。已知 TICs 可通过表达免疫抑制分子(如 CD155)及重塑肿瘤微环境(TME)实现免疫逃逸,但其在 HCC 中的具体机制及靶向 TICs 以增强免疫治疗效果的策略仍有待深入解析。本研究通过单细胞多组学分析及临床前模型,系统揭示 HCC 中 TICs 的免疫逃逸机制,并探索靶向 TICs「免疫特权」特性以增强抗 PD-1 治疗 efficacy 的新策略。
研究路线
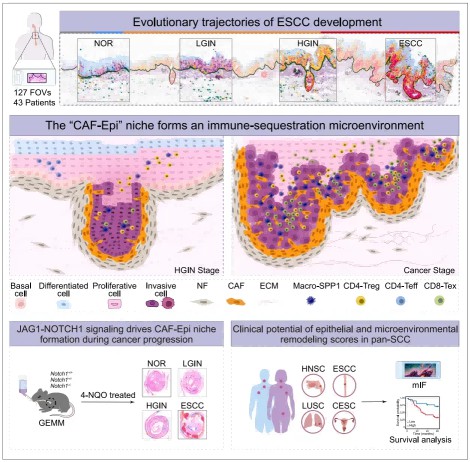
研究结论
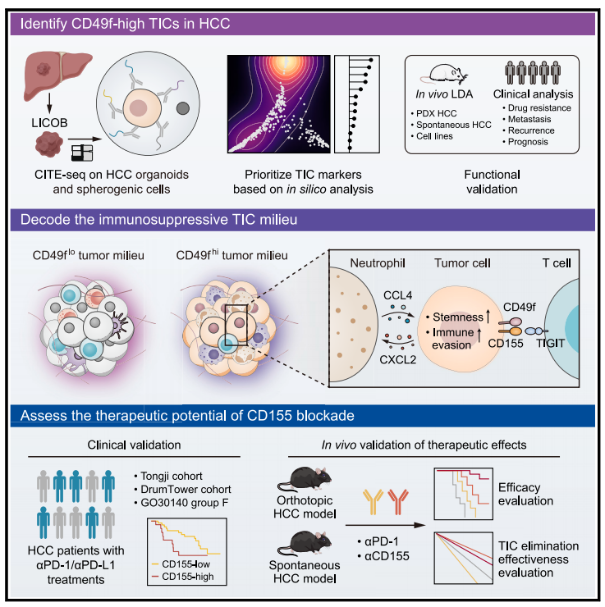
图 1. 肝癌类器官进行 CITE-seq 分析结果图
研究者对 LICOB 生物库来源的肝癌类器官进行 CITE-seq 分析,系统评估 14 个经典 TIC 标志物及 5 个免疫抑制相关表面标志物的表达特征,发现不同标志物在样本间存在显著表达异质性,且其标示 TIC 的能力差异显著。其中,CD49f(由 ITGA6 基因编码的细胞表面整合素蛋白)表现出最低的表达异质性及功能异质性,提示其作为 HCC 中 TIC 高效标志物的潜力(此前其在肝癌起始细胞中的应用价值尚未明确)。
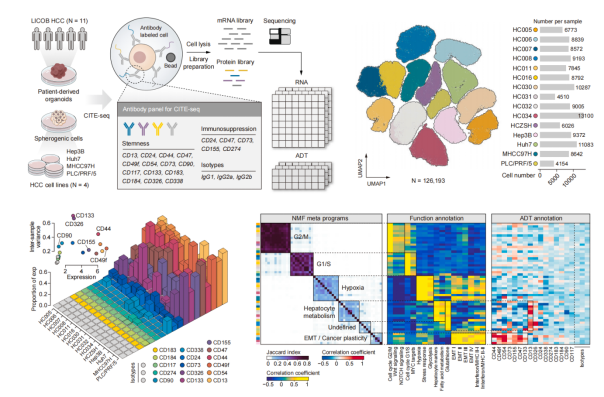
图 2. 利用单细胞多组学解析肿瘤异质性图
功能研究显示,CD49f 高表达的肝癌细胞可分泌大量髓系细胞招募相关因子,且与中性粒细胞存在密切互作:通过 SOX9 驱动的 CXCL2 分泌招募中性粒细胞,而中性粒细胞通过 CCL4-STAT3 轴维持并增强 TIC 特性。CCL4-STAT3 轴的激活可上调 PVR(CD155 编码基因)的表达,同时 CD49f 可通过与 CD155 在细胞膜上的直接相互作用稳定其蛋白水平。在上述双重机制作用下,CD49f<sup>high</sup>肝癌细胞通过高表达 CD155 获得「免疫特权」,从而逃避 CD8<sup>+</sup>T 细胞介导的免疫攻击。
研究发现,这种「免疫特权」同时构成该细胞亚群的「免疫弱点」。基于机制解析,研究者提出联合靶向 PD-1 与 CD155 的治疗策略,在临床前模型中证实该策略不仅可有效清除 TICs,还能减少 TME 中免疫抑制性中性粒细胞的浸润,显著增强抗 PD-1 治疗的效果。
研究意义
本研究通过单细胞多组学及临床前模型,揭示了 HCC 中 TICs 的新型免疫逃逸机制:明确 CD49f 作为 HCC 中 TIC 的高效标志物,其高表达的 TICs 通过 CXCL2-CXCR2 轴招募中性粒细胞,并经 CCL4/STAT3 信号通路诱导 CD155 表达,同时通过 CD49f 与 CD155 的蛋白互作增强其稳定性,最终获得免疫豁免特性以逃避免疫监视。该发现不仅为 HCC 免疫治疗提供了 CD155 这一新靶点,更揭示了联合靶向 CD155 与 PD-1/PD-L1 的潜在治疗价值——通过阻断 CD155 可显著增强抗 PD-1 治疗的 efficacy,为改善 HCC 患者治疗效果及预后提供了全新的理论依据与干预方向。
案例二:单细胞和空间转录组学整合分析揭示非小细胞肺癌联合治疗后的肿瘤微环境重塑特征

发表期刊:Molecular Cancer
影响因子:27.7
发表时间:2025 年 4 月
研究疾病:非小细胞肺癌(NSCLC)
样本类型:手术切除的肿瘤组织(新鲜组织、FFPE 样本)
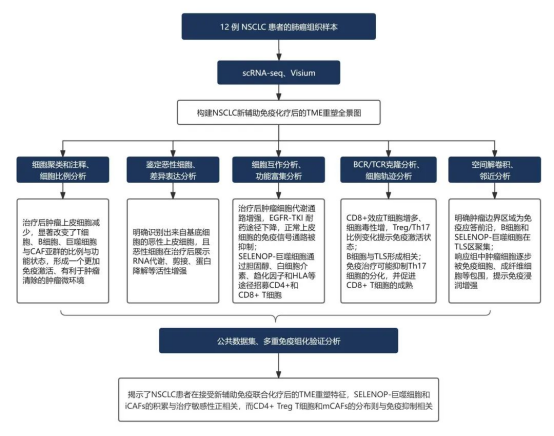
样本数量:12 例 NSCLC 患者队列的 16 份肺癌组织样本
样本分组:6 例未接受任何新辅助治疗的肺癌患者(TN 组),6 例接受 PD-L1 抑制剂联合顺铂化疗的患者(PT 组,包括 1 例 pCR(病理完全缓解),2 例 MPR(主要病理缓解),3 例 NMPR(非主要病理缓解)
应用技术:scRNA-seq、Visium、mIHC、IHC
研究背景
非小细胞肺癌(Non-small cell lung cancer,NSCLC)是最常见的肺癌类型,免疫检查点抑制剂(ICB)联合化疗已成为 NSCLC 新辅助治疗的一线方案。尽管部分患者可获得病理完全缓解,但疗效个体差异明显,免疫耐药机制仍不清晰。现有研究多聚焦于单细胞免疫图谱,缺乏肿瘤微环境(TME)空间异质性的深入理解。为全面解析新辅助治疗对 TME 的重塑作用,本研究通过单细胞与空间转录组联合策略,系统分析新辅助免疫治疗联合化疗对 NSCLC 患者肿瘤微环境的影响,旨在揭示与治疗响应相关的关键细胞亚群、分子特征及空间结构变化。
研究路线:
研究结论:
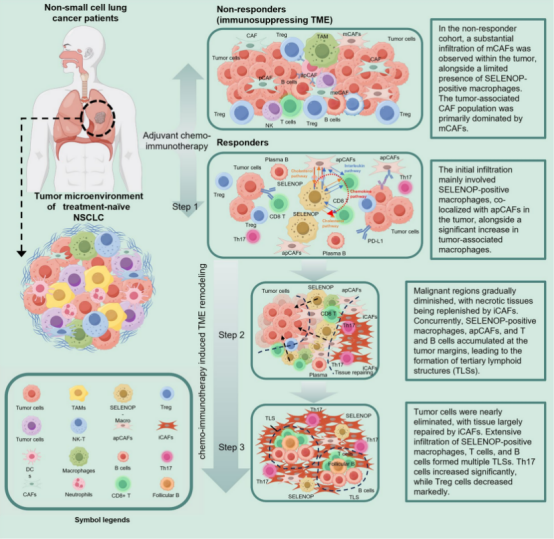
图 3. 影响 NSCLC 新辅助免疫化疗响应的关键细胞亚群及标志物图
通过和 scRNA-seq 和 Visium 空间转录组技术,研究者鉴定出影响 NSCLC 新辅助免疫化疗响应的关键细胞亚群及标志物。在治疗响应者中,观察到 CD4+Th17 细胞和炎症性癌相关成纤维细胞(iCAFs)比例显著上升,而 CD4+ Treg 细胞和基质性癌相关成纤维细胞(mCAFs)富集于非响应者,提示 Th17/Treg 和 iCAF/mCAF 比值或可作为治疗敏感性的生物标志物。同时,空间转录组显示 pCR 患者样本的肿瘤微环境(TME)中形成了富集 NK-T 细胞和 B 细胞的三级淋巴结构(TLS),与良好预后相关。
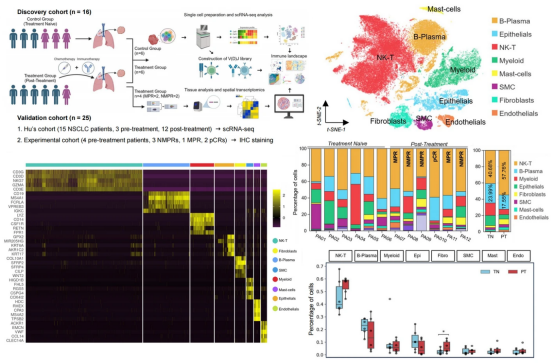
图 4. 使用单细胞测序解析 NSCLC 新辅助免疫化疗响应的关键细胞亚群图
进一步研究发现,SELENOP-巨噬细胞在治疗响应区域显著聚集,并在肿瘤边界处与抗抗原呈递癌相关成纤维细胞(apCAFs)之间存在强共定位。基于空间转录组的细胞间通信分析显示,SELENOP-巨噬细胞、apCAFs、CD4+和 CD8+T 细胞在空间上形成互作网络,且 SELENOP-巨噬细胞通过胆固醇、白细胞介素、趋化因子和 HLA 等途径招募 CD4+ Naïve、Helper 和 CD8+ Naïve T 细胞,从而增强免疫反应。
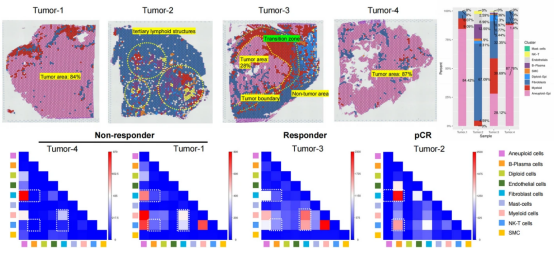
图 5. 使用空间转录组解析 NSCLC 新辅助免疫化疗后的空间微环境图
此外,治疗还引发了上皮细胞转录状态和功能的明显改变。在恶性上皮细胞中,治疗诱导细胞代谢、RNA 剪接、蛋白质水解等相关通路显著增强,同时 EGFR-TKI 耐药相关信号下调,提示治疗可能增强 TKI 敏感性;而在正常上皮细胞中,免疫相关通路(如 JAK-STAT、IL-17、抗原呈递)普遍抑制,一些关键的细胞周期和代谢通路(如 PI3K-Akt 信号通路、mTOR 信号通路和 Hippo 信号通路)也被抑制,表明治疗诱导了正常上皮细胞的免疫抑制和功能耗竭状态。
研究意义
本研究通过联合单细胞和空间转录组技术,系统揭示了 NSCLC 患者在接受新辅助免疫联合化疗后的肿瘤微环境重塑特征,识别出与治疗响应密切相关的关键细胞亚群、信号通路和空间结构特征,特别是 Th17/Treg 比例、SELENOP-巨噬细胞与 apCAFs 的协同作用以及 TSL 的形成。研究不仅弥补了以往对 TME 空间异质性认知的不足,也为解析免疫耐药机制和优化免疫联合治疗策略提供了新思路。此外,相关标志物和互作网络的发现具备重要的转化潜力,为个体化精准治疗提供了理论依据和潜在靶点。