实验操作篇
QIAGEN
1. 质粒载体构建不成功(目的片段和载体不能成功连接)的常见原因分析
①DNA 片段纯度差:体系中的杂质会降低连接效率,如果使用不纯化直接连接的方法一直无法成功构建载体,可以考虑进行纯化后再进行连接;
②目的片段和载体片段使用比例悬殊,或者用量过多或过少:目的片段和载体的摩尔比需要控制在一定范围之内,否则会影响连接效率,具体比例可以草靠连接酶说明书;
③连接条件不当:比如连接时间不足、温度不合适、酶失活等,可以通过设置对照组来排查原因;
④引物设计问题:比如通过无缝克隆的方式进行连接,同源臂区域如果 GC 含量过高过低,或者有重复序列,都可能在核酸外切酶切割后形成发卡结构,影响重组效率,可以使用质粒设计软件来检查同源重组臂的设计。
⑤插入序列本身的问题:有些序列本身存在连接载体困难、连接产物不稳定、连接产物在大肠杆菌中复制困难等问题。对于这类序列可以考虑将序列打断后分步构建,或者更换载体或感受态的方式尝试解决。
2. 质粒提取量不足
质粒提取量少常见的原因可能是:
①摇菌时间过长:大肠杆菌生长已经超过对数期,细菌老化,导致细胞和 DNA 降解。
②摇菌时间不足:细菌生长不充分,不能得到足够的菌量来抽提质粒,摇菌时间一般 12-14 小时为宜。
③质粒属于低拷贝类型,导致收集的细菌量过少。
④提取时菌量过多:容易导致裂解不充分,且容易超过硅胶膜柱的承载上限,最终使得质粒提取效率降低,建议参考提取试剂盒建议的菌液量来提取。
⑤质粒提取实验操作不当:比如在加入缓冲液重悬菌体时,菌体没有充分分散悬浮,导致后续裂解不充分。为指示菌体是否充分裂解,可以考虑在重悬缓冲液中加入指示剂(QIAGEN 质粒提取试剂盒中均有配备 LyseBlue 指示剂),均匀蓝色指示悬浮充分。
⑥试剂使用或存放不当:质粒提取试剂盒成分复杂且保存条件不同,有的试剂还容易出现浑浊。在使用之前需要检查试剂是否保存得当,如果出现浑浊,可参考试剂盒说明书,进行 37℃ 温浴至溶液澄清,再冷却至室温使用。
⑦洗脱液体积不合适:洗脱液使用体积过大会导致提取浓度偏低,过小会导致洗脱不充分,产量过低。建议参考试剂盒说明书来确定洗脱体积,如果洗脱浓度偏低,可以重复洗脱一次;
⑧洗脱液未添加到硅胶膜中心:可能会导致洗脱液覆盖不完全,导致洗脱效率降低。此外洗脱液可以使用碱性洗脱缓冲液或者无核酸酶的水,避免使用 PH<7 的缓冲液来洗脱;洗脱液提前预热至 65℃ 也可以提高洗脱效率。
3. 提取质粒转化大肠杆菌效率低
大肠杆菌转化效率受到多种因素的影响,例如感受态细胞状态,转化方式及操作(电转或热激),质粒用量,质粒质量等等。从感受态角度优化,可以考虑感受态本身的状态,比如贮存时间、贮存方式不当会导致感受态转化效率明显降低,此外热激条件也会影响感受态转化效率,建议热激时间严格按照感受态类型或说明书操作。从质粒角度优化,可以进一步确认质粒用量,质粒浓度,质粒是否降解等。
4. 提取质粒转染细胞效率低
质粒转染细胞的效率受到多种因素的影响,如细胞的类型,细胞的状态,转染时细胞的密度,转染的方式(电转或化转),DNA 的质量、转染试剂的选择等。从质粒角度来看,超螺旋比例高,且内毒素残留低的质粒会更有利于提高细胞转染的效率。
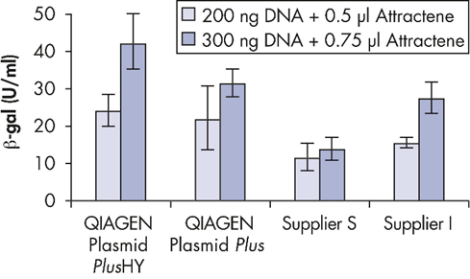
pCMVβ DNA 用不同试剂盒提取,转染 Huh-7 细胞,转染效率对比
5. 质粒酶切有问题,切不开或者酶切有杂带
酶切切不开可能跟酶本身状态、酶切缓冲液、酶切温度以及质粒本身都有关系。从酶切条件考虑,在确保酶活性的前提下,选择合适的酶切缓冲液、温度和时间;从质粒的角度考虑,需要确定内切位点的序列是否发生突变以及质粒中是否存在酶切抑制进而影响酶的活性,例如质粒提取时未将乙醇、盐分去除干净,此外 TE buffer 中的 EDTA 都有可能造成酶切抑制。
酶切质粒后发现条带弥散或者出现多个杂带,有可能是酶「切过了」,如酶切时间太长,或者酶量太多,可以尝试减少用量以及缩短酶切时间。此外,也有可能是质粒本身质量问题,例如质粒本身有降解,质粒的超螺旋比例很低,质粒中有残留的基因组 DNA,或者 RNA 等,甚至质粒中有二聚体或者多聚体,在电泳检测时都可能检测到一些非目标条带。可以通过电泳检测酶切之前的质粒判断质粒的质量,排除可能的原因。
调整酶切温度、时间、buffer 体系等,可以解决酶切杂带或切不开的问题。
6. PCR 扩增不出条带/条带弱/非特异条带怎么办?
①检查模板 DNA 是否存在降解,此外模板的上样量过高或者过低,也会影响片段的扩增;
②重新设计引物,比如引物与非目标序列同源性高可导致非特异扩增或无扩增产物,引物自身形成发夹结构、引物二聚体从而影响模板结合等。可以借助专业性软件检查引物结构并重新设计引物,比如 Primer Premier 5、NCBI Primer-BLAST、OligoCalc 等;
③调整 PCR 反应体系,PCR 酶失活或性能较差、Mg2+等组分浓度过高或过低都可能导致扩增失败,尤其是面对复杂片。建议选择高性能的 PCR 酶,提高 PCR 实验成功率。QIAGEN 的 AllTaq 系列所用的 Taq DNA 聚合酶利用小分子锁扣(Guard molecular)原理,大大提高片段扩增的灵敏度及特异性;
④调整 PCR 程序,退火温度过高、延伸时间过短、循环数过少都会导致扩增出现问题。建议首次拿到引物时,对模板浓度、Tm 值进行梯度测试,最终摸索出合适的扩增条件;
解决方案:
首先检查阳性对照(如已知成功扩增的模板 + 引物),排除试剂和仪器问题;如果阳性对照正常,逐步排查以下条件:模板质量→引物特异性→反应体系(Mg²⁺、酶)→程序参数(退火温度、循环数);针对复杂模板,可尝试选择高品质 PCR 酶、 「梯度测试」(模板浓度、退火温度),快速定位最优条件。
7. 胶回收效率低?如何提高?
胶回收效率低是分子克隆中常见的问题,可能与电泳操作、切胶、溶胶、结合、洗涤及洗脱等多个环节相关。可以从以下几个角度进行优化:
①选择合适的琼脂糖凝胶浓度:100-500bp 用 2%,500-2000bp 用 1.5%,2000-10000bp 用 1%,确保 DNA 条带清晰且易于释放。
②避免长时间紫外照射:尽量使用 302nm 长波长紫外灯,从切胶到放入溶胶 buffer 控制在 30 秒,也可选择蓝光染料及蓝光切胶仪。
③溶胶要彻底:严格按试剂盒比例添加溶胶 buffer,随后 50-60℃ 水浴加热溶胶,期间每隔 2-3 分钟颠倒混匀一次,直至胶块完全溶解。
④注意胶回收试剂盒内的回收柱吸附最大容量 ,避免 DNA 上样量过大导致超出柱子承载能力;
⑤充分洗涤,避免杂质残留:杂质成分会抑制 DNA 洗脱;
⑥不同试剂盒对特定长度的 DNA 回收率不同,例如 QIAquick Gel Extraction Kit 可以可纯化得到 70 bp 至 10 kb 的 DNA,回收效率高达 95%;MinElute PCR Purification Kit 可以洗脱纯化 70bp-4kb 的 DNA 产物。
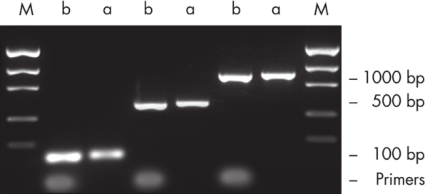
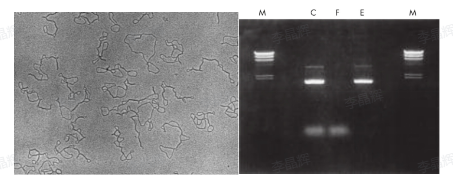
图为使用 MinElute PCR Purification Kit 纯化 PCR 产物对比,其中 b 为纯化前 PCR 产物,a 为纯化后产物,M 为 marker。
8. 感受态细胞效率不高?如何制备/选择/保存
制备:
感受态细胞的制备核心步骤包括菌体培养、低温处理、钙离子/电击诱导和分装冻存。实验过程中需注意无菌及低温操作,且需要在大肠杆菌中加入冷冻保护剂(如 10%-15% 甘油或 DMSO),其作用是降低冰点、减少冰晶形成,从而保护感受态细胞的转化效率;
选择:
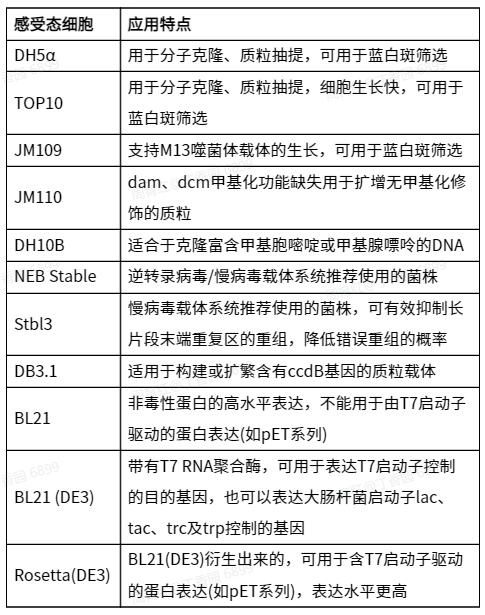
常见的大肠杆菌感受态细胞有多种,它们具有不同的基因型,通过基因型信息可以判断目的质粒是否可以使用这个感受态细胞来进行扩增,以及是否可以进行目的实验。例如 DH5α基因型:F-, φ80dlacZΔM15, Δ(lacZYA-argF )U169, deoR , recA1 , endA1 , hsdR17 (rK-, mK+), phoA, supE44 , λ-, thi-1, gyrA96 , relA1。基因型信息中有 lacZΔM15,说明其表达产物可以与 PUC 质粒编码表达的β-半乳糖苷酶的α肽段实现α互补,因此可以选择用 DH5α感受态来扩增 PUC 质粒并且做「蓝白斑」筛选。
保存:
①为避免反复冻融,建议小体积分装制备好的感受态(如 100μL / 管),分装所用的 EP 管需提前灭菌并预冷(4℃ 或 - 20℃),减少细胞温度波动;
②感受态细胞制备后,需立即用液氮或干冰 - 乙醇浴快速冷冻(1-2 分钟内降至 - 80℃ 以下),冻结后再转移至 - 80℃ 冰箱;
③-80℃ 低温保存,最大限度降低细胞代谢和冰晶形成对细胞膜的损伤,通常能稳定保存 6-12 个月(不同菌株略有差异),保存期间保证温度稳定,避免反复冻融;
内容来源:QIAGEN 凯杰