


- 询价
- 分子对接是一种基于生物信息学的模拟方法,它评估分子之间的相互作用,并通过计算机平台预测它们的结合模式和亲和力。分子对接常用于研究药物和受体相互作用的模型,特别适用于受体的作用机制、空间配置等研究得较清楚的体系。近年来,分子对接方法已成为计算机辅助药物研究领域的一项重要技术。
- 全国
- 2026年01月06日

企业认证
相关产品推荐更多 >





万千商家帮你免费找货
0 人在求购买到急需产品
- 详细信息
- 询价记录
- 文献和实验
- 技术资料
- 提供商:
湖北佰莱博科技有限公司
- 服务名称:
分子对接技术服务
- 规格:
1次
佰莱博分子对接(molecular docking)技术服务
一、技术简介
分子对接(molecular docking)是分子模拟的重要方法之一,其本质是两个或者多个分子之间通过几何匹配和能量匹配相互识别的过程。其中,几何匹配是分子间发生相互作用的基础,通过计算机计算出配体与受体最佳的空间结合构象;能量匹配则是分子间保持稳定结合的基础,通过对受体-配体复合物间的弱相互作用(包括氢键、范德华力、π-π相互作用等)进行计算,找到受体-配体复合物结合能最低,也就是最稳定的构象。
通过几何匹配和能量匹配,分子对接方法在小分子药物设计、多肽类药物设计、抗体类药物设计以及核酸抑制剂等领域有着广泛的应用。
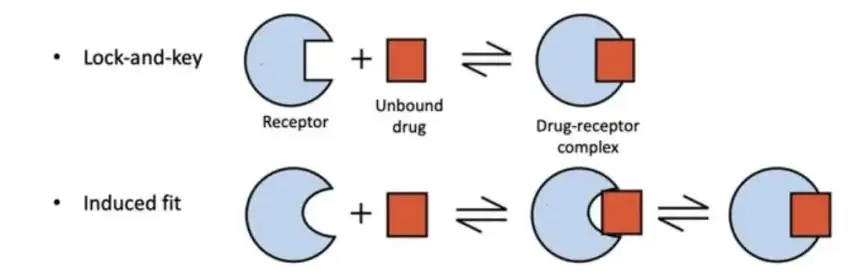
图1 分子对接原理简化示意图
二、服务内容
1. 同源建模,适用于蛋白无晶体结构的情况;
2. 蛋白和配体结构准备
3. 蛋白活性位点预测
4. 分子对接;
5. 从对接结果中,挑选结合能最低且复合物结构最稳定的结果进行可视化。
6.数据解读
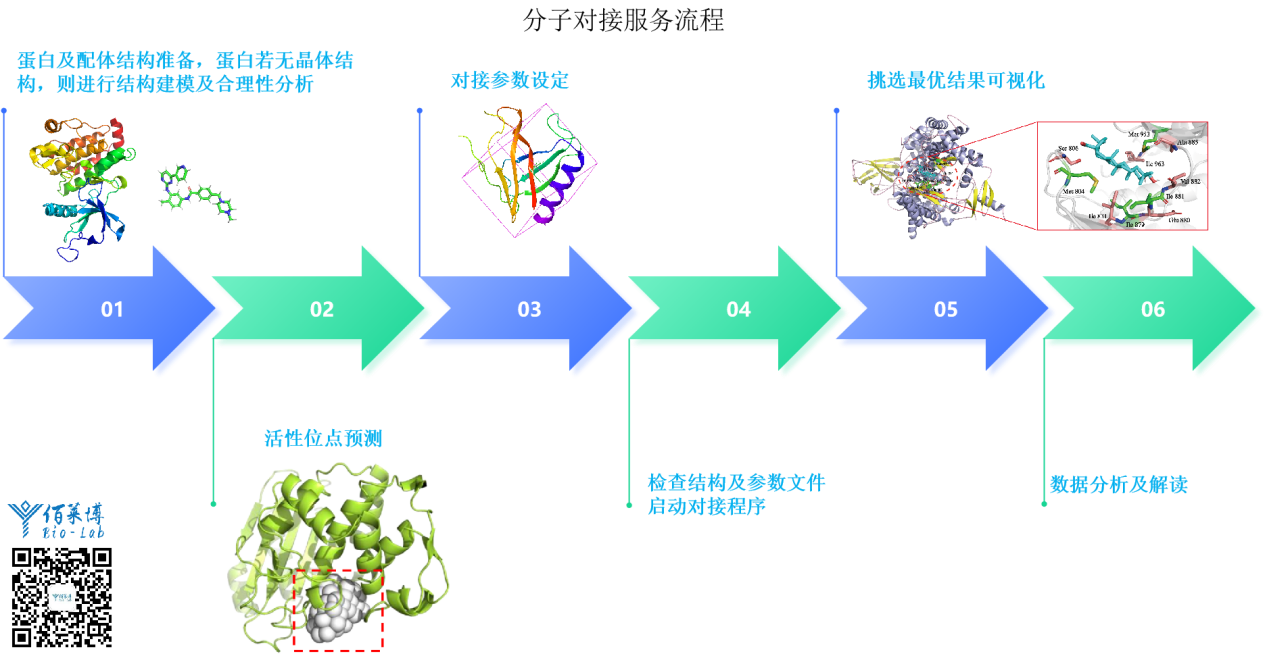
图2 分子对接技术流程图
三、技术参数
1、 受体信息:
① 需要提供PDB数据库蛋白编号;
② 需要提供Uniprot数据库蛋白/基因编号;
③ 蛋白的物种信息以及完整的氨基酸序列;
④ 蛋白的活性位点信息;
⑤ 相关的参考文献。
2、 配体信息:
① 如果配体是蛋白同受体信息要求;
② 普通小分子提供CAS号或者SMILES;
③ 普通小分子也可以直接提供结构(建议Chemdraw软件绘制);
④ 普通小分子尽量提供2D结构文件(sdf/mol2等常用格式);
3、 活性位点信息:
提供的受体蛋白没有活性位点信息需要额外的AI位点预测服务。
四、结果分析
随着X-RAY晶体衍射和NMR技术的进步,越来越多的蛋白质的晶体结构被解析。这些晶体结构并不能直接用于分子对接时,有时解析的结构信息会包含一些错误,如氨基酸缺失等等,在对接前需要对蛋白质的结构进行修复,才能用于后续的分子对接。一般而言解析的晶体结构缺失氢原子的位置信息,在对接前需要进行加氢、计算电荷分布才能用于对接。
准备蛋白结构后需要寻找药物分子结合的活性位点,针对靶标蛋白的活性位点进行分子对接能极大地提高分子对接的准确性。而靶标蛋白的活性位点信息可以通过已报道的文献研究来获得,如果确实没有相关的信息,也可以通过计算机预测,寻找合适的活性位点。
亲和力的单位为kcal/mol(AutodockVina),即受体与配体相互结合的强度。一般认为当这个能量的值为负值时,受体与配体的结合能自发进行,同时亲和力的绝对值越大表明受体-配体复合物的稳定性越高。
在对对接结果分析时,除了关注亲和力的大小,也应当检查受体与配体之间的相互作用,非键弱相互作用( 氢键 ≈ π-cation键 > π-π堆积 > 范德华力)对于维持受体-配体复合物的稳定十分重要。在选择最佳的对接结果时,不应当只考虑亲和力的大小,更应该关注受体与配体之间形成的弱相互作用。
五、结果交付
1、位点预测图
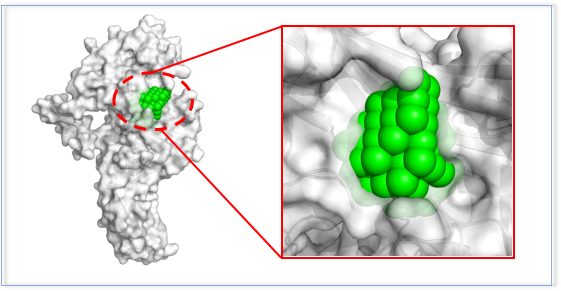
预测的蛋白的活性位点
表1 预测的结合口袋处的蛋白残基
|
作用残基 |
作用残基 |
作用残基 |
作用残基 |
|
LYS 12 |
THR 129 |
VAL 168 |
ASP 298 |
|
ARG 26 |
VAL 143 |
ILE 169 |
THR 299 |
|
ARG 63 |
ASP 144 |
ARG 170 |
PHE 391 |
|
GLU 67 |
PHE 145 |
ASP 291 |
ALA 392 |
|
ASP 68 |
GLU 146 |
GLY 292 |
ARG 393 |
|
LEU 69 |
LYS 161 |
LYS 293 |
|
|
SER 127 |
ARG 166 |
LYS 295 |
|
|
VAL 128 |
LEU 167 |
HIS 296 |
|
2、所有对接可能构象亲和力打分表
|
结合模式 |
亲和力(kcal/mol) |
距最佳结果的RMSD(l.b.) |
距最佳结果的RMSD(u.b.) |
|
1 |
-5.395 |
0 |
0 |
|
2 |
-4.874 |
16.484 |
19.772 |
|
3 |
-4.859 |
17.186 |
19.848 |
|
4 |
-4.832 |
2.193 |
6.121 |
|
5 |
-4.742 |
2.475 |
5.915 |
|
6 |
-4.732 |
1.949 |
4.624 |
|
7 |
-4.633 |
18.786 |
22.312 |
|
8 |
-4.591 |
3.088 |
6.88 |
|
9 |
-4.557 |
19.724 |
22.888 |
3、优势结合构象对接图绘制
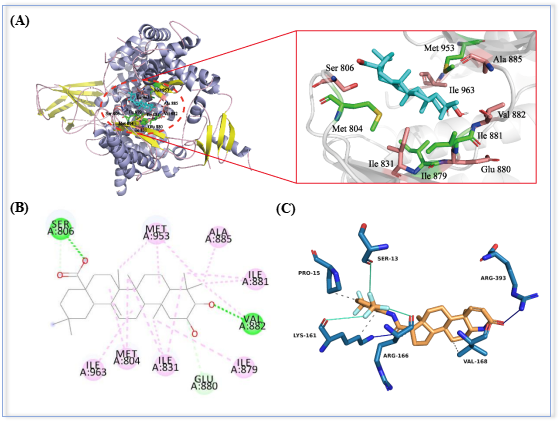
六、 服务项目
分子对接:
使用Autodock vina对受体和配体的结合模式进行预测,并计算亲和力,初步筛选与受体相互作用的配体。可进一步进行分子动力学模拟,评估复合物的稳定性。
分子动力学模拟技术服务:
通过gromacs对蛋白与小分子化合物的对接结果进行100ns动力学模拟,判断复合物的稳定性,辅助验证蛋白与小分子化合物相互作用的关键氨基酸位点。
虚拟筛选技术服务:
小分子化合物库与蛋白的对接,筛选得到化合物亲和力排序,可以挑选排名靠前的做SPR等体外分子互作技术高通量验证。
反向找靶技术服务:
根据客户提供的小分子化合物,在人、小鼠、大鼠靶点库中找到相匹配的蛋白靶点,给出蛋白打分列表,后期进行分子对接和亲和力检测实验验证。
对接图补充
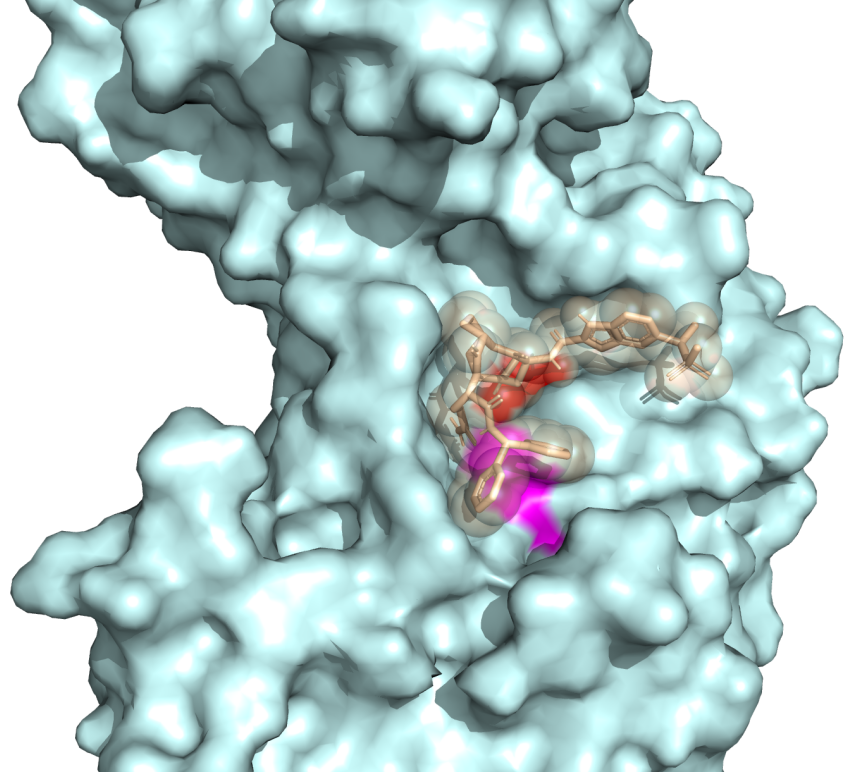
结合表面图
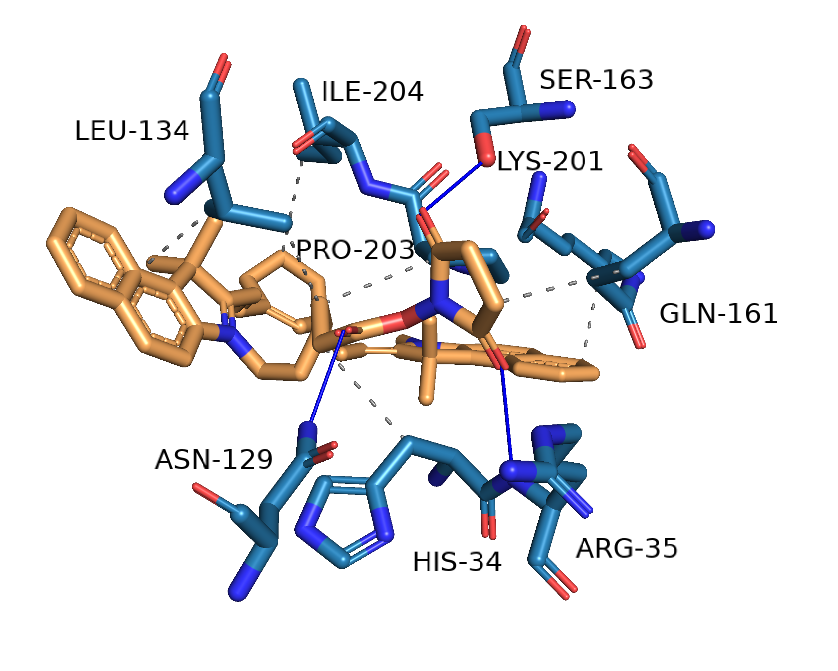
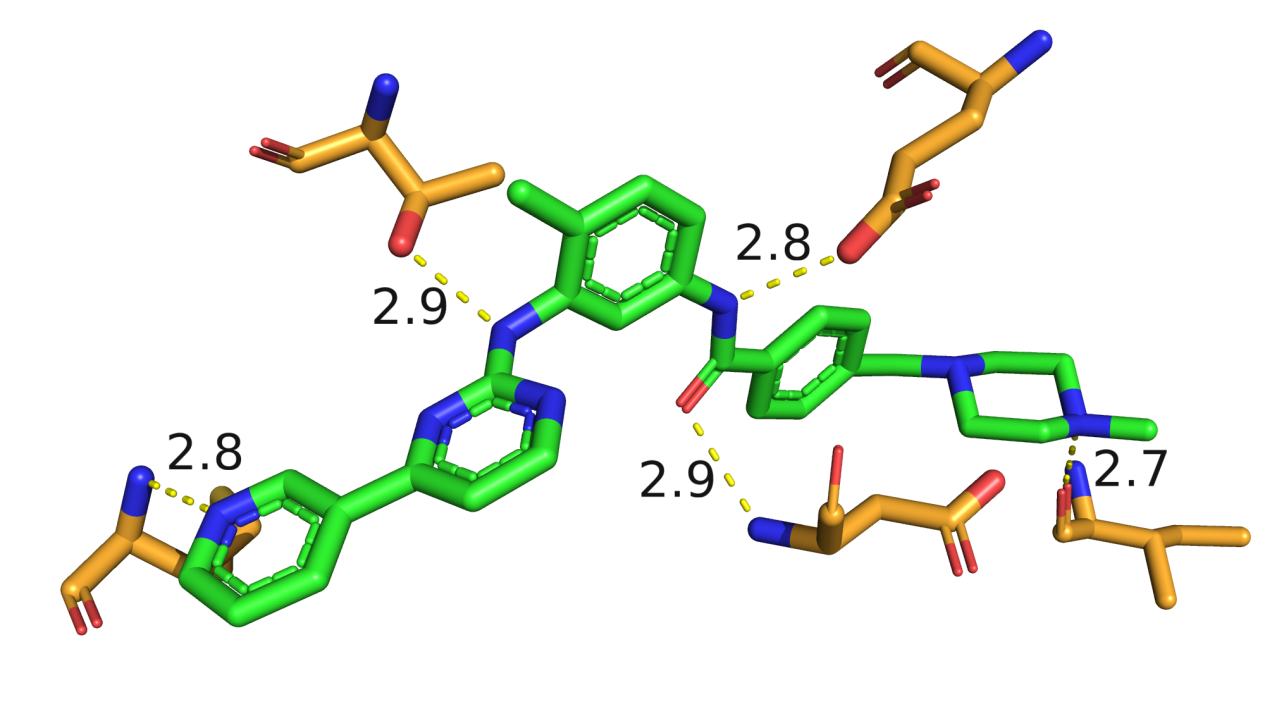
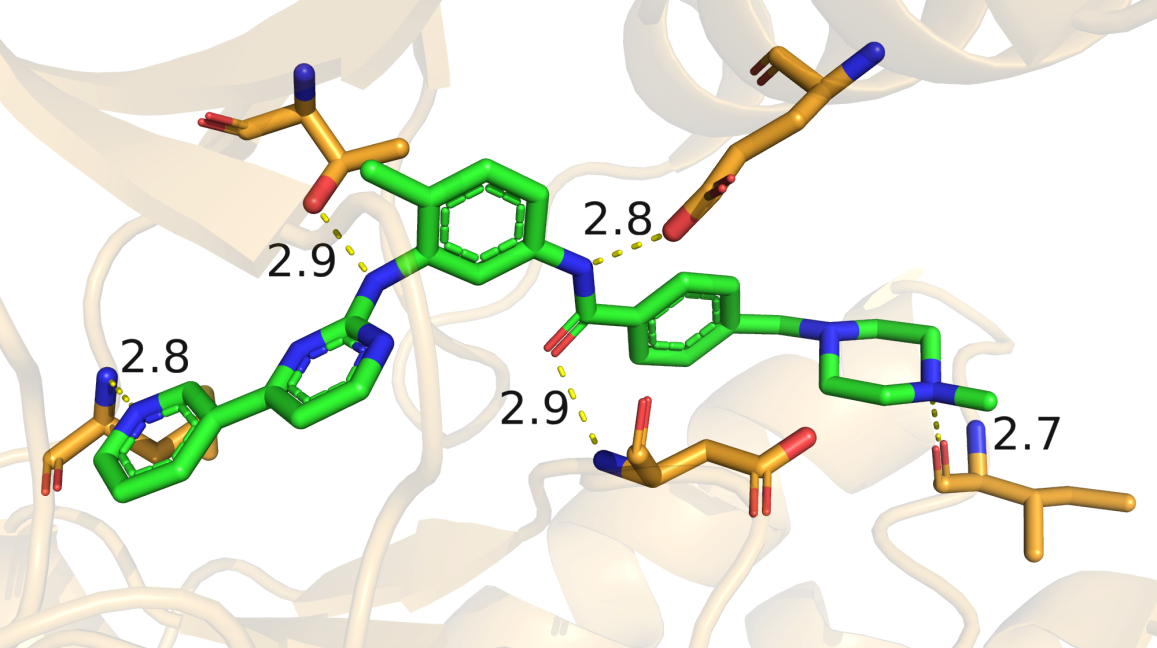
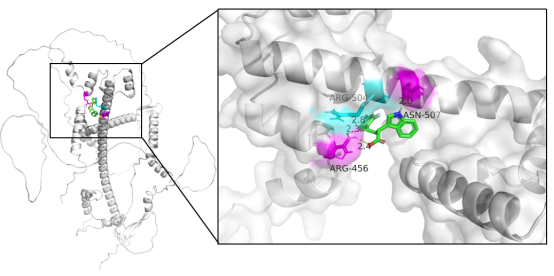
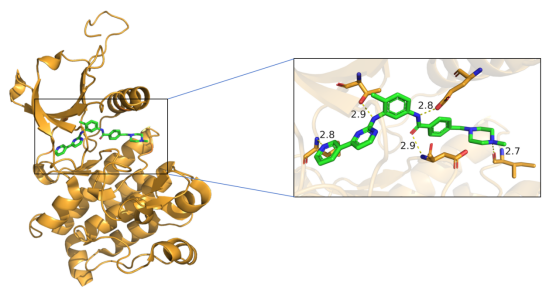
相互作用图
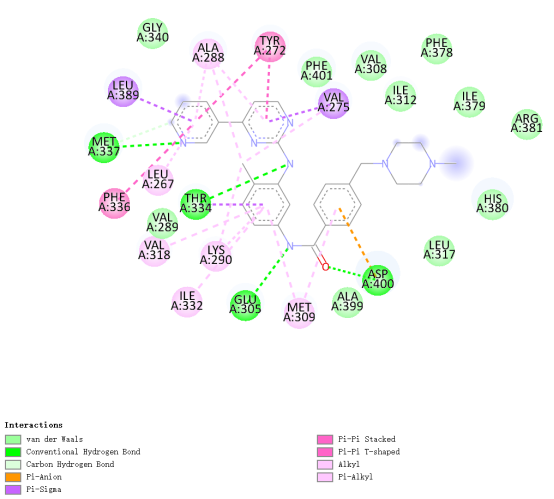
2D相互作用图
佰莱博生物提供计算机模拟、生物物理、生物化学及细胞生物学等多个层面分子互作筛选及验证服务。
风险提示:丁香通仅作为第三方平台,为商家信息发布提供平台空间。用户咨询产品时请注意保护个人信息及财产安全,合理判断,谨慎选购商品,商家和用户对交易行为负责。对于医疗器械类产品,请先查证核实企业经营资质和医疗器械产品注册证情况。
- 作者
- 内容
- 询问日期
 文献和实验
文献和实验Molecular docking is a key tool in structural molecular biology and computer-assisted drug design. The goal of ligand—protein docking is to predict the predominant binding mode(s) of a ligand with a protein of known three-dimensional
Molecular Docking Methodologies
Molecular docking represents an important technology for structure-based drug design. Docking is a computational technique aimed at the prediction of the most favorable ligand–target spatial configuration and an estimate of the corresponding
Flexible Ligand Docking with Glide
degrees of freedom are available to determine the optimal ligand orientation relative to a rigid protein receptor geometry. This unit presents protocols for flexible ligand docking with Glide, optionally including ligand constraints or ligand molecular
 技术资料
技术资料暂无技术资料 索取技术资料
