


- 询价
- 中国
- 人源化模型
- 2026年04月07日

企业认证
相关产品推荐更多 >





万千商家帮你免费找货
0 人在求购买到急需产品
- 详细信息
- 文献和实验
- 技术资料
- 服务名称:
基因敲入模型
- 提供商:
赛业生物
B6-hSMN2(SMA)小鼠
产品编号:C001504
品系全称:C57BL/6NCya-Smn1tm1(hSMN2)/Cya
品系背景:C57BL/6NCya
传代建议:杂合与杂合互配
本产品为HUGO-GT (Humanized Genomic Ortholog for Gene Therapy) 系列小鼠
品系描述
脊髓性肌萎缩症(Spinal Muscular Atrophy, SMA)是由脊髓前角细胞运动神经元变性引起的常染色体隐性遗传病,以严重进行性肌无力和肌萎缩为特征,影响控制人体进行呼吸、爬行、走动和头颈控制以及吞咽等活动的肌肉,进而增加患者罹患肺炎和呼吸道感染的风险。SMA是婴幼儿期最常见的致死性神经遗传病,人群发病率为1/6,000~1/10,000,是罕见病中的常见疾病之一。
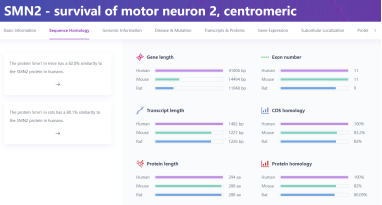
SMN1基因编码的生存运动神经元(SMN)蛋白是真核细胞生物生存所必需的管家蛋白,负责运动神经元存活的维持,SMN1双等位基因致病性突变可导致SMA的发生。人体中存在和SMN1基因高度同源SMN2基因,两者仅存在几个碱基序列的差异,SMN2基因在7号外显子剪接增强子处存在(c.840C>T)突变使SMN2 Pre-mRNA的可变剪接模式与SMN1不同,导致生成的多数SMN2 mRNA缺少第7个外显子,编码功能缺失的SMN截断蛋白,并在细胞内迅速降解 [1]。仅有部分SMN2 Pre-mRNA(10%~15%)能被剪切为全长mRNA,编码具有正常功能的SMN蛋白。约95%的SMA患者携带SMN1第7号外显子纯合缺失突变或使SMN1转为SMN2的突变,SMN2表达无法代偿体内SMN蛋白的缺失,导致疾病的发生。目前已获批的药物主要通过补充SMN1基因或靶向调节SMN2选择性剪切来治疗SMA。SMN2靶向治疗通过改变其剪切方式以增加SMN全长蛋白的表达。小鼠作为最普遍的临床前实验对象,只存在Smn1基因且Smn1纯合敲除致死,因此,构建出可模拟人类SMA致病机理且符合人类疾病进程的小鼠模型对靶向药物等多种疗法的开发和验证至关重要,全人源化动物模型的应用有助于推动SMA相关的潜在疗法向临床试验进一步转化。
本模型为SMN2基因人源化模型,利用基因编辑技术将小鼠内源性Smn1基因替换为人源SMN2基因片段,可在小鼠体内模拟SMA患者的致病机理。在该模型中,小鼠Smn1基因被人源SMN2基因取代,由于SMN2基因主要产生7号外显子缺失的SMNΔ7蛋白而非全长SMN蛋白,人源化SMN2基因无法完全弥补Smn1缺失所导致的异常,使模型表现为SMA样表型。此外,由于不同亚型的SMA与SMN2拷贝数相关,本模型可以与在6号染色体中插入SMN2基因的Rosa26-hSMN2小鼠交配来增加小鼠体内SMN2拷贝数,以提升模型的生存周期,并模拟不同SMA亚型,可用于更多相关的致病机理和药物临床前研究。
罕见病数据中心(RDDC)
(1) 基因基本信息
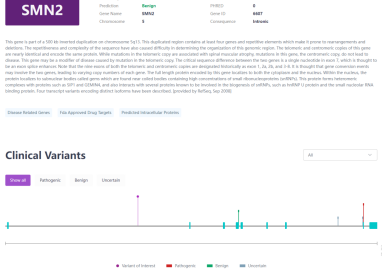
(2) 临床突变信息
(3) 疾病介绍
脊髓性肌萎缩症(Spinal Muscular Atrophy,SMA)是一种常染色体隐性遗传病,其特征是脊髓前角运动神经元受损,导致进行性肌无力和肌萎缩,可影响控制人体进行呼吸、爬、走、头颈控制以及吞咽等活动的肌肉,进而增加患者罹患肺炎和呼吸道感染的风险。该疾病是婴幼儿期最常见的致死性神经遗传病,人群发病率为1/6,000~1/10,000。SMN蛋白的表达量与疾病的严重程度相关,SMA以SMN2的拷贝数为依据可被分为五种表型,其中0型患者SMN2拷贝数最少,病情最为严重,IV型症状最轻。目前在中国约有3万名SMA患者,致病变异的携带率约为1/50。
(4) 基因及突变介绍
SMN1基因是SMA的重要致病基因,其编码的SMN蛋白是真核细胞生物生存所必需的管家蛋白,维持运动神经元的存活,而SMN1双等位基因发生致病性突变可引发SMA。人基因组中存在高度同源的SMN1基因和SMN2基因。SMN1基因和SMN2基因仅存在几个碱基的差异,但SMN2基因7号外显子中携带的一个关键碱基差异(c.840C>T)使SMN2 pre-mRNA的可变剪接与SMN1基因不同,导致SMN2基因主要产生7号外显子缺失且不稳定的SMNΔ7蛋白[1]。绝大多数SMA患者为SMN1基因突变,而高度同源的SMN2基因不能产生足量的全长SMN蛋白以弥补SMN1基因功能的缺失,进而导致疾病发生。
在SMA患者中,约95%的病例是由于SMN1基因第7号外显子纯合缺失所致(0+0型),通常伴有第8号外显子的缺失;约5%的病例是由于SMN1基因复合杂合突变所致(0+1d型),即一个等位基因发生缺失,另一个等位基因发生微小致病性变异;极少数患者则是由于SMN1双等位基因发生微小致病性变异所导致(1d +1d型)[2]。
(5) 基因治疗
目前SMA的治疗有多种策略,第一种通过靶向SMN2改变其剪切方式,以增加SMN全长蛋白的表达量。小鼠作为最普遍的临床前实验对象,只存在Smn1基因且Smn1纯合敲除致死,构建出模拟人类SMA致病机理且符合人类疾病进程的小鼠模型对靶向SMN2药物等多种疗法的开发和验证至关重要,全人源化动物模型的应用有助于推动SMA相关的潜在疗法向临床试验进一步转化。IONIS公司开发的诺西那生钠注射液(Spinraza)是一种反义寡核苷酸(ASO)药物,其临床前中使用了Δ7小鼠,这是最为经典的SMA小鼠模型。该小鼠在敲除鼠源Smn1基因的基础上,通过转基因技术转入了人SMN2全长基因以及敲除7号外显子的SMN的cDNA(SMN2 delta7)[3]。该三重纯合子小鼠在出生时的体型明显小于正常同窝对照鼠,并表现出进行性肌肉无力,平均生存期为17.7天,Spinraza靶向SMN2基因7号内含子的剪切位点,对SMN2 Pre-mRNA剪切模式进行修饰,使其生成大量包含7号外显子的正常SMN2 mRNA以编码功能性SMN蛋白,使Δ7小鼠的寿命得到延长 [4]。第二种为补充SMN1基因的替代疗法,由腺相关病毒载体(AAV)递送SMN1基因到患者体内,可弥补SMN1基因突变导致的蛋白表达缺失。由诺华研发的Zolgensma正是此类药物,该药物已经获批上市。
此外,CK-2127108和SRK-015等作用于肌肉的小分子药物以及干细胞移植疗法等都在进行研发中。
(6) 模型总结
综上所述,SMN1基因是SMA的重要致病基因,开发拷贝数确定且能稳定遗传的SMA疾病模型对于SMA的相关研究至关重要。赛业生物研发的SMN2全基因人源化模型不仅具有与SMA患者相似的疾病同源性和行为表象,而且拷贝数确定,能够稳定遗传,是SMA相关基因治疗中更优的临床前研究疾病模型。
发布时间:2024-03-28
- Confidential -
风险提示:丁香通仅作为第三方平台,为商家信息发布提供平台空间。用户咨询产品时请注意保护个人信息及财产安全,合理判断,谨慎选购商品,商家和用户对交易行为负责。对于医疗器械类产品,请先查证核实企业经营资质和医疗器械产品注册证情况。
 文献和实验
文献和实验The spinal muscular atrophies are a clinically and genetically heterogeneous group of neuromuscular disorders caused by degeneration of anterior horn cells. In proximal spinal muscular atrophy (SMA), the muscles of the extremities closest
三句话读懂一篇 CNS:睡眠太多或太少,都会增加中风风险;筛选高质量精子,科学家开发了新方法
缩症(SMA)是一种严重的遗传性神经肌肉疾病,生存期不超过 2 周岁。 2023 年 3 月 30 日,碱基编辑技术开创者刘如谦(David Liu)教授团队在 Science 杂志发表研究论文 Base editing rescue of spinal muscular atrophy in cells and in mice。 该研究使用双 AAV 载体递送的腺嘌呤碱基编辑器(ABE),经过一次性治疗,将脊髓型肌萎缩症(SMA)小鼠模型的运动能力恢复到接近正常的水平,并显著地延长了其寿命,且安全有效
Application of Viral Vectors to Motor Neuron Disorders
Motor neuron diseases such as amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA) are characterized by the progressive loss of motor neurons in the spinal cord and primary motor cortex. Subsequent paralysis of skeletal
 技术资料
技术资料暂无技术资料 索取技术资料

