


- 询价
- 中国
- 2026年04月02日

企业认证
相关产品推荐更多 >


万千商家帮你免费找货
0 人在求购买到急需产品
- 详细信息
- 文献和实验
- 技术资料
- 提供商:
赛业生物
产品编号:C001504
品系全称:C57BL/6NCya-Smn1tm1(hSMN2)/Cya
品系背景:C57BL/6NCya
传代建议:杂合与杂合互配
品系描述
脊髓性肌萎缩症(Spinal Muscular Atrophy, SMA)是由脊髓前角细胞运动神经元变性引起的常染色体隐性遗传病,以严重进行性肌无力和肌萎缩为特征,影响控制人体进行呼吸、爬行、走动和头颈控制以及吞咽等活动的肌肉,进而增加患者罹患肺炎和呼吸道感染的风险。SMA是婴幼儿期最常见的致死性神经遗传病,人群发病率为1/6,000~1/10,000,是罕见病中的常见疾病之一。
SMN1基因编码的生存运动神经元(SMN)蛋白是真核细胞生物生存所必需的管家蛋白,负责运动神经元存活的维持,SMN1双等位基因致病性突变可导致SMA的发生。人体中存在和SMN1基因高度同源SMN2基因,两者仅存在几个碱基序列的差异,SMN2基因在7号外显子剪接增强子处存在(c.840C>T)突变使SMN2 Pre-mRNA的可变剪接模式与SMN1不同,导致生成的多数SMN2 mRNA缺少第7个外显子,编码功能缺失的SMN截断蛋白,并在细胞内迅速降解 [1]。仅有部分SMN2 Pre-mRNA(10%~15%)能被剪切为全长mRNA,编码具有正常功能的SMN蛋白。约95%的SMA患者携带SMN1第7号外显子纯合缺失突变或使SMN1转为SMN2的突变,SMN2表达无法代偿体内SMN蛋白的缺失,导致疾病的发生。目前已获批的药物主要通过补充SMN1基因或靶向调节SMN2选择性剪切来治疗SMA。SMN2靶向治疗通过改变其剪切方式以增加SMN全长蛋白的表达。小鼠作为普遍的临床前实验对象,只存在Smn1基因且Smn1纯合敲除致死,因此,构建出可模拟人类SMA致病机理且符合人类疾病进程的小鼠模型对靶向药物等多种疗法的开发和验证至关重要,全人源化动物模型的应用有助于推动SMA相关的潜在疗法向临床试验进一步转化。
本模型为SMN2基因人源化模型,利用基因编辑技术将小鼠内源性Smn1基因替换为人源SMN2基因片段,可在小鼠体内模拟SMA患者的致病机理。在该模型中,小鼠Smn1基因被人源SMN2基因取代,由于SMN2基因主要产生7号外显子缺失的SMNΔ7蛋白而非全长SMN蛋白,人源化SMN2基因无法完全弥补Smn1缺失所导致的异常,使模型表现为SMA样表型。此外,由于不同亚型的SMA与SMN2拷贝数相关,本模型可以与在6号染色体中插入SMN2基因的Rosa26-hSMN2小鼠交配来增加小鼠体内SMN2拷贝数,以提升模型的生存周期,并模拟不同SMA亚型,可用于更多相关的致病机理和药物临床前研究。
构建方式
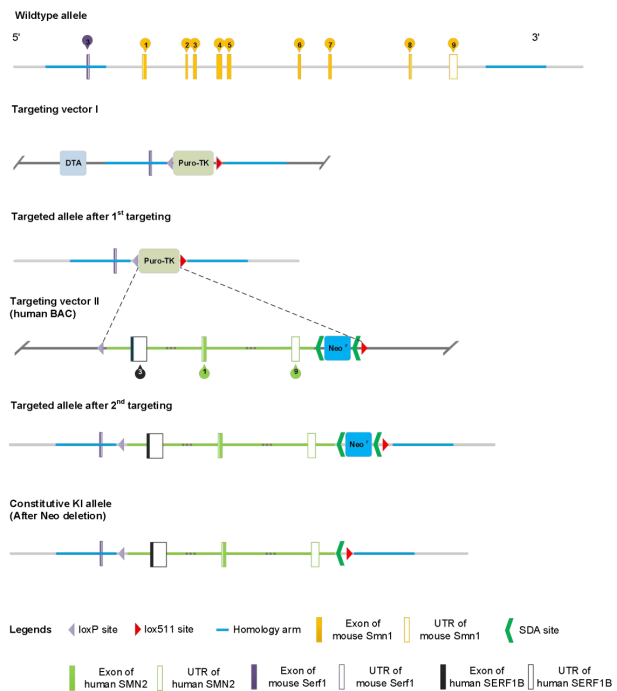
图1. B6-hSMN2(SMA)小鼠基因编辑打靶示意图。利用胚胎干细胞(ES)基因编辑打靶技术,将小鼠Smn1基因上游10kb至下游0.5kb的片段替换为人源SMN2基因上游15kb至下游5kb片段。
研究应用
B6-hSMN2(SMA)小鼠可用于脊髓性肌萎缩症(SMA)致病机制和治疗药物的临床前评价等研究。
验证数据
- 人源SMN2基因和鼠源Smn1基因表达检测
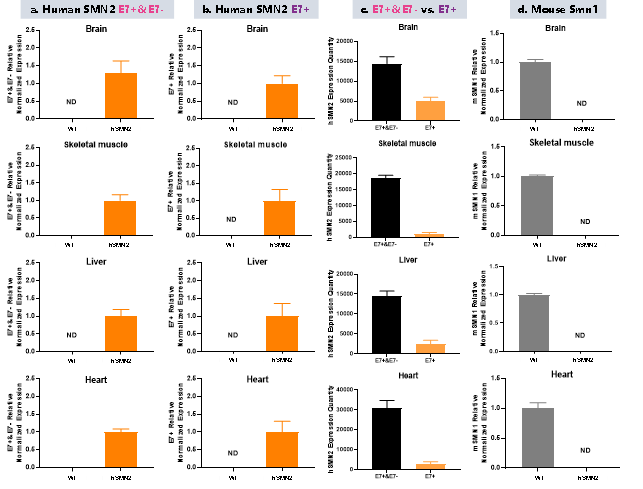
图2. 3周龄雌性野生型小鼠(WT)和纯合B6-hSMN2(SMA)小鼠(hSMN2)脑部、肝脏、心脏和骨骼肌中人源SMN2基因和小鼠Smn1基因表达的检测(n=3)。通过RT-qPCR检测人源SMN2基因和小鼠Smn1基因的表达以及包含7号外显子的SMN2转录本(E7+)比例。结果显示,与野生型小鼠相比,B6-hSMN2(SMA)小鼠脑部、肝脏、心脏和骨骼肌中均存在人源SMN2基因的表达,小鼠Smn1基因的表达缺失。B6-hSMN2(SMA)小鼠体内包含7号外显子(E7+)的转录本仅占总人源SMN2转录本(E7+&E7-)的极少部分。
E7+&E7-:总人源SMN2转录本;E7-:缺失7号外显子的人源SMN2转录本;E7+:包含7号外显子的人源SMN2转录本;ND:未检测到(Not detected)。
- SMN蛋白表达检测
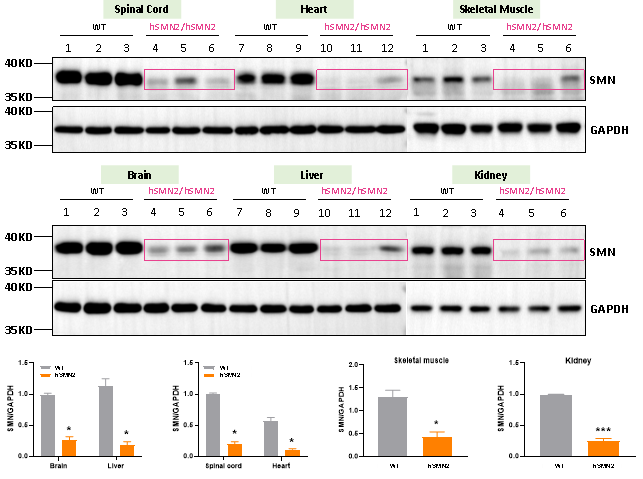
图3. 3周龄雌性野生型小鼠(WT)和纯合B6-hSMN2(SMA)小鼠(hSMN2/hSMN2)脊髓、心脏、骨骼肌、脑部、肝脏和肾脏SMN蛋白的表达。通过Western Blot检测WT小鼠与B6-hSMN2(SMA)小鼠中SMN蛋白的表达水平。结果显示,纯合hSMN2小鼠脊髓、心脏、骨骼肌、脑部、肝脏和肾脏SMN蛋白表达均严重下调,表明该小鼠体内仅存在少量由SMN2基因编码的SMN蛋白。
- B6-hSMN2(SMA)小鼠的生存曲线
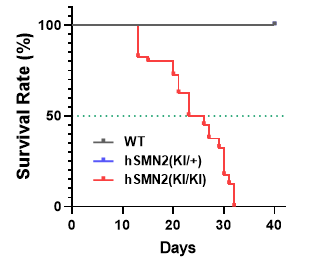
图4. 野生型小鼠(WT)和B6-hSMN2(SMA)小鼠(hSMN2)的生存率。结果显示,纯合B6-hSMN2(SMA)小鼠在13天左右开始出现死亡,20天左右达半数致死率,在40天前小鼠基本全部死亡。而杂合hSMN2小鼠与野生型相似,暂无生存异常,符合SMA常染色体隐性遗传的特性。
- B6-hSMN2(SMA)小鼠的外观
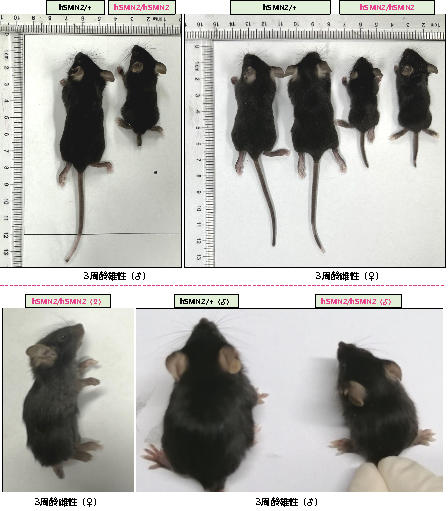
图5. 纯合和杂合B6-hSMN2(SMA)小鼠在3周龄时的外观。与同窝杂合B6-hSMN2(SMA)小鼠(hSMN2/+)相比,纯合B6-hSMN2(SMA)小鼠(hMSN2/hSMN2)表现出肌肉萎缩,站立不稳,体型较小,身长较短,断尾,四肢浮肿的现象。
- B6-hSMN2(SMA)小鼠肌肉组织学病理
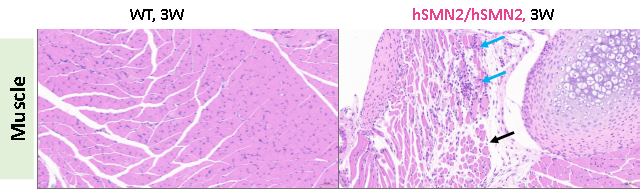
图6. 3周龄雌性纯合B6-hSMN2(SMA)小鼠(hSMN2/hSMN2)和野生型小鼠(WT)肌肉组织H&E染色。B6-hSMN2(SMA)小鼠肌肉组织灶性可见少量肌细胞坏死,胞质崩解,伴有极少量的淋巴细胞浸润(蓝色箭头所示),周围可见肌细胞萎缩,体积减小,肌细胞间隔增宽、排列疏松(黑色箭头所示),肌间神经无异常。
- B6-hSMN2(SMA)小鼠脚掌组织学病理
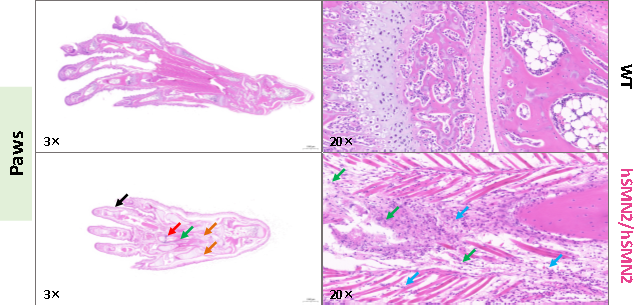
图7. 3周龄雄性纯合B6-hSMN2(SMA)小鼠(hSMN2/hSMN2)和野生型小鼠(WT)脚掌组织H&E染色。B6-hSMN2(SMA)小鼠(hSMN2)脚趾各处关节结构清晰,周围偶见游离粒细胞浸润(黑色箭头所示)。脚掌周围局部可见肌纤维坏死溶解,结构消失并多见结缔组织增生取代原有的肌纤维(如绿色箭头所示),伴有多量的粒细胞浸润(蓝色箭头所示),偶见掌骨断裂(红色箭头所示)。多见皮下水肿,结缔组织疏松,间隙增宽并伴有少量的粒细胞浸润(橙色箭头所示)。
- B6-hSMN2(SMA)小鼠尾部组织学病理
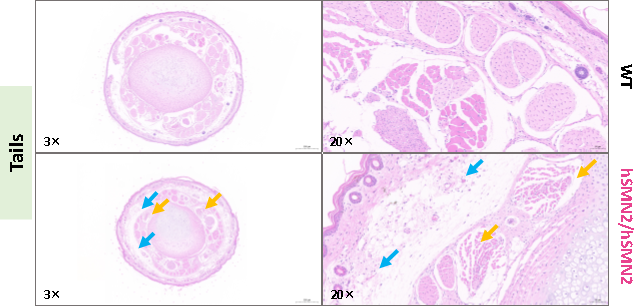
图8. 3周龄雌性纯合B6-hSMN2(SMA)小鼠(hSMN2/hSMN2)和野生型小鼠(WT)尾部组织H&E染色。B6-hSMN2(SMA)小鼠(hSMN2)尾部组织表皮及真皮无明显异常,局部皮下水肿,结缔组织排列疏松,偶见血管扩张,伴有极少量的淋巴细胞浸润(蓝色箭头所示)。与对照组相比,肌层多见肌细胞萎缩,体积减小(黄色箭头所示);组织中央为尾椎骨,无明显异常。
- 靶向SMN2小核酸药物提升B6-hSMN2(SMA)小鼠SMN蛋白表达并增加脊髓前角运动神经元个数
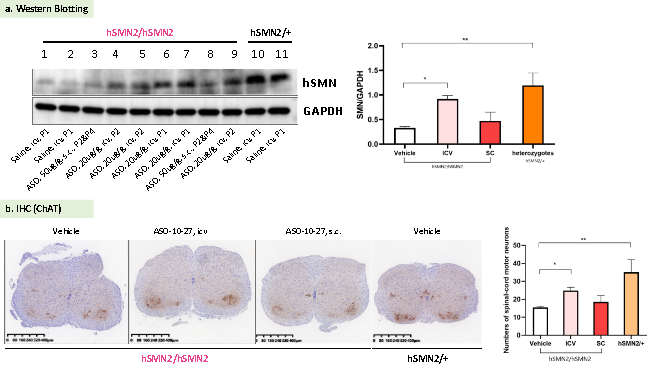
图9. 靶向调节SMN2剪切模式的ASO分子对纯合B6-hSMN2(SMA)小鼠(hSMN2/hSMN2)的影响。以治疗SMA的ASO药物Spinraza*为基础,合成结构和功能类似的反义寡核苷酸ASO-10-27(由GenScript合成)。然后分别通过脑室内注射(icv)和皮下注射(s.c.)的方式,给B6-hSMN2(SMA)小鼠分别注射不同剂量的ASO-10-27。结果显示,脑室内注射的ASO可以提高B6-hSMN2(SMA)小鼠脑部的SMN蛋白表达量(如图10. a所示),并增加脊髓前角的运动神经元数量(如图10. b所示)。
*Spinraza为首/款获批的SMA治疗药物,通过反义寡核苷酸(ASO)对SMN2 Pre-mRNA剪切模式进行修饰,使其生成大量包含7号外显子的正常SMN2 mRNA以编码功能性SMN蛋白 [4]。
- 靶向SMN2小核酸药物减轻B6-hSMN2(SMA)小鼠疾病表型并提高生存率
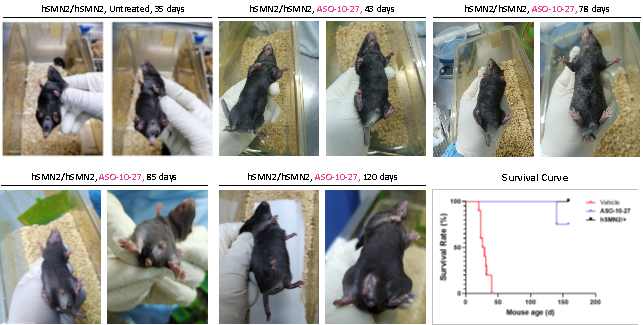
图10. ASO分子提升纯合B6-hSMN2(SMA)小鼠(hSMN2/hSMN2)存活率并延缓组织病变。经ASO-10-27治疗后,纯合B6-hSMN2(SMA)小鼠的生存率显著提高,所有接受ASO处理的小鼠在140日龄时才开始死亡。相比之下,未接受ASO-10-27处理的B6-hSMN2(SMA)小鼠的半数存活期仅为29天,35日龄时就出现脚趾坏死和断尾现象,到40日龄左右全部死亡。然而,接受ASO治疗的B6-hSMN2(SMA)小鼠在43日龄时只出现脚趾轻微肿胀,且尾巴完好,未见脚趾坏死。到78日龄,只有少数小鼠出现断尾现象,脚趾仍未坏死。85日龄时,虽有部分小鼠断尾,但脚趾依然健康。直到120日龄,部分小鼠才出现脚掌坏死的现象。这些结果表明ASO-10-27治疗能显著改善B6-hSMN2(SMA)小鼠的生存状况和健康状况。
罕见病数据中心(RDDC)
- 基因基本信息
- 临床突变信息
- 疾病介绍
脊髓性肌萎缩症(Spinal Muscular Atrophy,SMA)是一种常染色体隐性遗传病,其特征是脊髓前角运动神经元受损,导致进行性肌无力和肌萎缩,可影响控制人体进行呼吸、爬、走、头颈控制以及吞咽等活动的肌肉,进而增加患者罹患肺炎和呼吸道感染的风险。该疾病是婴幼儿期最常见的致死性神经遗传病,人群发病率为1/6,000~1/10,000。SMN蛋白的表达量与疾病的严重程度相关,SMA以SMN2的拷贝数为依据可被分为五种表型,其中0型患者SMN2拷贝数最少,病情最为严重,IV型症状最轻。目前在中国约有3万名SMA患者,致病变异的携带率约为1/50。
- 基因及突变介绍
SMN1基因是SMA的重要致病基因,其编码的SMN蛋白是真核细胞生物生存所必需的管家蛋白,维持运动神经元的存活,而SMN1双等位基因发生致病性突变可引发SMA。人基因组中存在高度同源的SMN1基因和SMN2基因。SMN1基因和SMN2基因仅存在几个碱基的差异,但SMN2基因7号外显子中携带的一个关键碱基差异(c.840C>T)使SMN2 pre-mRNA的可变剪接与SMN1基因不同,导致SMN2基因主要产生7号外显子缺失且不稳定的SMNΔ7蛋白[1]。绝大多数SMA患者为SMN1基因突变,而高度同源的SMN2基因不能产生足量的全长SMN蛋白以弥补SMN1基因功能的缺失,进而导致疾病发生。
在SMA患者中,约95%的病例是由于SMN1基因第7号外显子纯合缺失所致(0+0型),通常伴有第8号外显子的缺失;约5%的病例是由于SMN1基因复合杂合突变所致(0+1d型),即一个等位基因发生缺失,另一个等位基因发生微小致病性变异;极少数患者则是由于SMN1双等位基因发生微小致病性变异所导致(1d +1d型)[2]。
- 基因治疗
目前SMA的治疗有多种策略,第一种通过靶向SMN2改变其剪切方式,以增加SMN全长蛋白的表达量。小鼠作为最普遍的临床前实验对象,只存在Smn1基因且Smn1纯合敲除致死,构建出模拟人类SMA致病机理且符合人类疾病进程的小鼠模型对靶向SMN2药物等多种疗法的开发和验证至关重要,全人源化动物模型的应用有助于推动SMA相关的潜在疗法向临床试验进一步转化。IONIS公司开发的诺西那生钠注射液(Spinraza)是一种反义寡核苷酸(ASO)药物,其临床前中使用了Δ7小鼠,这是最为经典的SMA小鼠模型。该小鼠在敲除鼠源Smn1基因的基础上,通过转基因技术转入了人SMN2全长基因以及敲除7号外显子的SMN的cDNA(SMN2 delta7)[3]。该三重纯合子小鼠在出生时的体型明显小于正常同窝对照鼠,并表现出进行性肌肉无力,平均生存期为17.7天,Spinraza靶向SMN2基因7号内含子的剪切位点,对SMN2 Pre-mRNA剪切模式进行修饰,使其生成大量包含7号外显子的正常SMN2 mRNA以编码功能性SMN蛋白,使Δ7小鼠的寿命得到延长 [4]。第二种为补充SMN1基因的替代疗法,由腺相关病毒载体(AAV)递送SMN1基因到患者体内,可弥补SMN1基因突变导致的蛋白表达缺失。由诺华研发的Zolgensma正是此类药物,该药物已经获批上市。
此外,CK-2127108和SRK-015等作用于肌肉的小分子药物以及干细胞移植疗法等都在进行研发中。
- 模型总结
综上所述,SMN1基因是SMA的重要致病基因,开发拷贝数确定且能稳定遗传的SMA疾病模型对于SMA的相关研究至关重要。赛业生物研发的SMN2全基因人源化模型不仅具有与SMA患者相似的疾病同源性和行为表象,而且拷贝数确定,能够稳定遗传,是SMA相关基因治疗中更优的临床前研究疾病模型。
参考文献
[1]Wirth B , Karakaya M , Kye M J , et al. Twenty-Five Years of Spinal Muscular Atrophy Research: From Phenotype to Genotype to Therapy, and What Comes Next[J]. Annual Review of Genomics and Human Genetics, 2020(1).
[2]Wirth B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA)[J]. Hum Mutat. 2000;15(3).
[3]Hill SF, Meisler MH. Antisense Oligonucleotide Therapy for Neurodevelopmental Disorders[J].Dev Neurosci. 2021;43(3-4)
[4]Mendell JR, Al-Zaidy S, Shell R, et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy[J].N Engl J Med. 2017 Nov 2;377(18):1713-1722.
风险提示:丁香通仅作为第三方平台,为商家信息发布提供平台空间。用户咨询产品时请注意保护个人信息及财产安全,合理判断,谨慎选购商品,商家和用户对交易行为负责。对于医疗器械类产品,请先查证核实企业经营资质和医疗器械产品注册证情况。
 文献和实验
文献和实验外观:因为高脂饮食,毛发看起来有些「油腻」。 活动:因 DIO 的肥胖表型,多数小鼠看起来比较「慵懒」,不爱动。 脱毛:因 B6 品系的特性,容易出现理毛导致的脱毛,需关注是否有继发皮肤炎症反应。 运输应激:DIO 小鼠在经历运输后容易出现体重降低的情况。根据友商资料和我们的历史数据,运输后 DIO 平均体重减轻在 10%-15%。一般需要经过 2 周左右的适应性饲养,体重可以恢复到出库水平。 打架现象:因雄鼠的天性问题,可能会出现打架的情况,建议不要将不同笼小鼠进行混装。 B6 小鼠会
年,来自 Jackson Laboratory 的 C.Ostermeier 等人对脱脂牛奶 / 棉子糖冷冻小鼠精子方法的修改,方法是在 CPA(3%脱脂奶和 18%棉子糖组合)中添加单硫代甘油(MTG),这种方法能让 C57BL/6J 品系受精率提高到 60%。近交系 B6 冷冻的方案得到了质的提升,也能满足冷冻需求。 2010 年,Takeo 和 Nakagata 又改良了冷冻方案,即在原来的组合基础上添加了 L-谷氨酰胺,L-谷氨酰胺是血浆和组织中含量最丰富的游离氨基酸,L-谷氨酰胺具有
、FVB 等。1. C57BL/6 小鼠也叫 Black 6 (B6)、C57 或 C57 Black 6,是使用最为广泛的近交系小鼠。毛色为黑色,是目前基因工程小鼠模型、人类疾病小鼠模型、自发或诱导突变品系最常用的遗传背景。2. BALB/c 小鼠毛色为白色,被大家熟知主要是由于其在单克隆抗体与免疫学研究中的贡献。BALB/c 小鼠对放射线极为敏感。乳腺癌发病率低,但随着年龄增长,患其他癌症(如肺癌和肾癌)的几率大为增加。3. FVB 小鼠毛色白色,起源于 Swiss 小鼠。该小鼠受精卵有大而显著
 技术资料
技术资料暂无技术资料 索取技术资料

