

11月Cell重磅:新线粒体细胞死亡方式!或将引爆国自然新热点!先天免疫和代谢信号通过线粒体周膜裂解诱导线粒体依赖性膜裂解
2025-12-10 10:46点击次数:290
关键词:先天性免疫激活和代谢紊乱的组合在许多疾病中起着关键作用,通常导致线粒体功能障碍和氧化应激,从而驱动发病机制。然而,在这些条件下的机制调控目前仍不明确。在此,作者报告了一种由先天性免疫信号传导和代谢紊乱诱导的独特溶解性细胞死亡机制,该机制不依赖于caspase活性以及先前描述的焦亡(pyroptosis)、泛凋亡(PANoptosis)、坏死性凋亡(necroptosis)、铁死亡(ferroptosis)和氧凋亡(oxeiptosis)。相反,经历BAX/BAK1/BID依赖性氧化应激的线粒体与质膜保持长时间接触,导致局部氧化损伤,作者将这一过程称为mitoxyperiosis(线粒体氧化损伤过程)。该过程进而引起膜溶解和细胞死亡,称为mitoxyperilysis(线粒体氧化溶解性死亡)。mTORC2调控这种细胞死亡,而mTOR抑制可恢复细胞骨架活性,使片状伪足(lamellipodia)回缩并将线粒体从膜上动员离开,从而维持膜的完整性。在体内激活该通路可在依赖mTORC2的方式下使肿瘤消退。总体而言,该研究结果确定了一种响应先天性免疫信号传导与代谢紊乱协同作用的溶解性细胞死亡方式。该研究于2025年11月发表于《Cell》上,影响因子42.5,题目为“Innate immune and metabolic signals induce mitochondria-dependent membrane lysis via mitoxyperiosis”。
技术路线

研究思路
1. 先天性免疫激活和代谢紊乱诱导炎症性细胞死亡

图1. 先天性免疫激活与代谢紊乱协同诱导炎症性细胞死亡和炎症小体激活
2. 响应协同性IIAMD的细胞死亡不依赖于焦亡、凋亡、坏死性凋亡、泛凋亡或铁死亡
NLRP3被认为可检测体内稳态改变以诱导炎症小体激活、gasdermin D(GSDMD)裂解和焦亡或泛凋亡。然而,NLRP3对IIAMD诱导的细胞死亡是可有可无的(图2A)。作者评估了关键成孔分子gasdermins的作用,发现尽管Gsdmc4、Gsdmd和Gsdme在BMDMs中高表达(图2B),但小干扰RNA(siRNA)介导的Gsdmc4敲低以及Gsdmd、Gsdme和坏死性凋亡执行者Mlkl的基因敲除并未阻止细胞死亡(图2C)。此外,GSDMD对IL-1β和IL-18的释放也是可有可无的(图1H)。最近被鉴定为介导质膜破裂和较大DAMPs释放的分子NINJ1,对细胞死亡也是可有可无的(图2D)。然而,如在其他细胞死亡方式中所见,NINJ1是质膜破裂所必需的,以允许LDH和HMGB1释放(图2E)。这些观察共同表明,IIAMD诱导的膜损伤和溶解性细胞死亡独立于众所周知的成孔执行者。
为了进一步深入了解机制,作者检测了细胞死亡分子的激活情况。caspase-1/3/7/8的裂解、GSDMD/GSDME的裂解以及MLKL的磷酸化,这些可作为凋亡、焦亡、泛凋亡和坏死性凋亡的标志物,在LPS加CS反应中被诱导(图S2A)。然而,泛caspase抑制剂z-VAD和坏死性凋亡抑制剂Nec-1s单独或联合使用均未能抑制细胞死亡(图2F),表明该细胞死亡独立于caspases,且不是凋亡、焦亡、泛凋亡或坏死性凋亡。响应LPS加CS的细胞死亡随z-VAD处理而增加(图2F),这与z-VAD加LPS可诱导强烈坏死性凋亡一致。铁死亡抑制剂Ferrostatin-1(Fer-1)单独或与z-VAD和Nec-1s联合使用均不能阻断细胞死亡(图2F)。关键调控因子Casp1、Casp11、Casp8和Ripk3的基因敲除,无论是否同时阻断残留caspase活性(z-VAD)、RIPK1活性(Nec-1s)和铁死亡激活(Fer-1),均不能提供保护(图2G)。作者研究了caspase-9的作用,鉴于其作为内在凋亡启动因子的已知功能。在Casp1−/−Casp11−/−Casp8−/−Ripk3−/−(QKO)BMDMs中敲低Casp9(图2H和2I)以及使用CRISPR敲除Casp9(图2J和2K)均无保护作用。综上所述,这些发现表明IIAMD诱导的细胞死亡独立于caspases和主要的已表征细胞死亡通路。
3. 谷胱甘肽耗竭和氧化应激导致协同性IIAMD下的细胞死亡
作者接下来试图通过无偏见的代谢组学方法表征这种先前未知的细胞死亡过程的代谢特征。与正常培养基、CS或LPS处理相比,LPS加CS处理的细胞具有独特的代谢谱(图S2D)。在LPS加CS组相对于其他组显著改变的大多数代谢物(231个中的209个)丰度降低(图S2E)。代谢通路分析显示LPS加CS组谷胱甘肽(GSH)代谢降低(图S2F),并通过正交方法进一步证实(图S2G)。
GSH是一种抗氧化剂,可对抗活性氧(ROS)并防止氧化损伤。鉴于ROS和氧化应激在介导细胞死亡中的核心作用,以及LPS加CS降低了GSH水平(图S2G),作者假设氧化应激在IIAMD诱导的细胞死亡中发挥关键作用。使用CellROX Deep Red(CellROX-DR)荧光探针对活细胞成像显示,在细胞膜损伤和细胞死亡之前,LPS加CS反应中细胞内氧化应激逐渐增加(图2L)。大多数碘化丙啶(PI)阳性细胞(表明其发生细胞死亡)也含有CellROX-DR斑点(图2L、2M和S2H),表明正在经历死亡的细胞中存在氧化应激。单独的LPS诱导了强烈的氧化应激(图2L、2M、S2H和S2I),但细胞死亡很少(图1A、1B和2M)。这可能是由于单独的LPS反应中GSH产生增加(图S2G),因为外源性GSH剂量依赖性地减少了LPS加CS反应中的细胞死亡(图S2J)。此外,除TLR3配体poly(I:C)外,大多数TLR配体和TNF在结合CS时均诱导氧化应激(图S2K),这与作者观察到的除TLR3配体外TLR配体结合CS时诱导细胞死亡一致(图1A、1B、1J、1K和S1K–S1M)。
接下来,作者试图确定增加氧化应激以驱动细胞死亡所需的上游信号传导。TNF加CS需要TNF受体信号传导(图S2L),而LPS加CS利用TLR4和MyD88诱导氧化应激(图S2M),这与它们对细胞死亡的需求一致(图1J和1K)。巯基化合物N-乙酰-L-半胱氨酸(NAC)降低了氧化应激(图S2M)并挽救了LPS加CS诱导的细胞死亡细胞(图2N),进一步将氧化应激与该细胞死亡机制联系起来。
先前的研究鉴定了氧凋亡(oxeiptosis),这是一种由氧化还原感受器KEAP1介导的caspase非依赖性、氧化应激诱导的细胞死亡通路。siRNA敲低Keap1并未减少LPS加CS反应中的细胞死亡(图S2N和S2O),表明KEAP1介导的氧凋亡不参与IIAMD诱导的细胞死亡。KEAP1抑制NRF2信号传导;然而,RNA测序(RNA-seq)分析显示LPS加CS富集了NRF2靶基因的表达(图S2P),进一步表明这种细胞死亡与氧凋亡不同。
总体而言,作者的数据表明IIAMD耗竭GSH,通过一种先前未知的通路引起氧化应激和细胞死亡。

图2. 对先天性免疫激活和代谢紊乱的细胞死亡反应依赖于氧化应激,独立于caspases,且不属于焦亡、凋亡、坏死性凋亡、铁死亡或泛凋亡
4.经历氧化应激的线粒体在细胞溶解前持续与质膜结合
作者在CellROX-DR染色的垂死细胞边缘观察到荧光"斑点"(图2L)。为了进一步理解氧化应激细胞器的亚细胞动态与IIAMD诱导的细胞死亡之间的关系,作者进行了共聚焦活细胞成像(图S3A;见视频S1)。在LPS加CS反应中,细胞内CellROX-DR阳性斑点在质膜附近稳定定位长时间(例如>20分钟)(图S3A;视频S1)。在斑点与膜接触部位,质膜最初退化,最终破裂,允许膜不通透性Sytox染料进入并染色细胞核(图S3A;视频S1)。
为了鉴定哪些细胞器发生氧化应激,作者将CellROX-DR与LysoView(一种染色溶酶体的溶酶体染料)或MitoView(一种染色线粒体基质的染料)共染色。CellROX-DR主要与MitoView共定位,而非LysoView(图3A),表明氧化应激定位于线粒体,与线粒体是ROS主要细胞内来源一致。线粒体ROS分析确实模拟了CellROX-DR的结果(图S3B和S3C)。使用MitoView的活细胞成像显示,在IIAMD反应中,线粒体在膜退化和细胞通透性形成前长时间靠近细胞膜(图3B–3F;见视频S2)。相比之下,在对照条件下,线粒体-膜共定位是短暂的(图3B–3D和3F;见视频S3、S4、S5和S6)。
为了定量评估线粒体-膜动态,作者开发了一个分析工作流程,通过时间追踪膜相关线粒体(图S3D;见视频S7)。虽然线粒体在所有测试条件下都活跃地与膜结合,但当细胞用培养基、CS或单独LPS处理时,大多数结合是短暂的(例如<4分钟)(图3G和3H)。相比之下,LPS加CS诱导了显著更长的结合,一些线粒体保持与膜结合延长的时间(例如>20分钟)(图3E、3G和3H)。这种延长的线粒体-膜接触是IIAMD诱导的细胞死亡特有的,因为用LPS加尼日利亚菌素处理的细胞表现出更短暂、瞬时的线粒体-膜接触(图3F–3H)。这些发现表明,延长的线粒体-膜接触与IIAMD诱导的细胞死亡之间存在独特联系。
5. 线粒体-膜接触位点的局部氧化损伤驱动细胞溶解
由于氧化自由基寿命短且主要由线粒体产生,作者假设延长的线粒体-膜结合可能促进局部膜氧化损伤,随后导致细胞破裂。为了在空间上检查线粒体-膜接触位点和氧化损伤,作者用4-羟基-(4-HNE)染色,这是一种脂质过氧化产物,可与赖氨酸、组氨酸或半胱氨酸形成加合物(4-HNE-K/H/C),作为氧化损伤的替代标志物。将4-HNE-K/H/C与F-actin和线粒体共染色,可定量外周线粒体(与F-actin共定位的线粒体)处的局部4-HNE-K/H/C强度。LPS加CS处理后,作者在线粒体共定位的细胞外周观察到增加的4-HNE-K/H/C信号,这些信号被NAC处理降低(图3I和S3E)。此外,作者通过BodipyC11测量观察到脂质过氧化水平增加(图S3F)。
为了进一步理解氧化损伤、线粒体和细胞膜之间的时间空间关系,作者在MitoTracker存在下进行了比例BodipyC11成像。与作者之前的成像结果一致(图3E),在线粒体发生形变之前,线粒体持续定位于细胞外周靠近膜破裂部位(图3J,Mitotracker通道)。外周脂质过氧化焦点也与线粒体共定位(图3J,氧化的BodipyC11和Merge通道)。在膜破裂前,每个位点的脂质过氧化逐渐增加(图3J;见视频S8)。透射电子显微镜(TEM)成像显示,与对照细胞相比,IIAMD处理的细胞在线粒体样细胞器(即基质肿胀)电子密度降低的区域附近,质膜受损部分存在损伤(图S3G)。
总之,这些数据表明IIAMD诱导延长的线粒体-膜接触,导致膜局部氧化损伤的积累,作者通过mitoxyperiosis(线粒体将ROS递送到细胞外周的过程)这一过程指代。该过程随后导致局部膜破裂和细胞裂解,即mitoxyperilysis。

图3. 线粒体被保留在质膜处以促进局部膜氧化损伤,并通过mitoxyperilysis驱动细胞死亡
6. BAX-、BAK1-和BID依赖性线粒体损伤和氧化应激是mitoxyperilysis的驱动因素
氧化还原失衡与线粒体功能障碍和损伤有关。为了进一步了解IIAMD期间线粒体的状态,作者首先测量了维持线粒体膜电位(ΔΨm)的线粒体呼吸作用。IIAMD损害了基础氧消耗率(OCR)和最大OCR(图S4A和S4B)。一致地,IIAMD降低了ΔΨm(图S4C),提示线粒体损伤。
为了检查线粒体损伤在mitoxyperilysis中的作用,作者专注于含BH3结构域的蛋白,因为它们具有破坏线粒体的功能。作者通过CRISPR/Cas9生成了单重、双重或三重缺陷的Bax、Bak1和Bid的永生化BMDM(iBMDM)敲除细胞系(图S4D)。Bax−/−Bak1−/−Bid−/−细胞受到保护免于IIAMD诱导的细胞死亡和炎症小体激活,其中大部分保护功能归因于BAX和BAK1(图S4E和S4F)。为了理解BAX、BAK1和BID如何促进细胞死亡,作者检查了线粒体损伤(通过测量ΔΨm)和氧化应激(通过评估GSH和线粒体ROS水平)。尽管对照细胞在LPS加CS反应中失去了ΔΨm,但Bax−/−Bak1−/−Bid−/−细胞具有更高的ΔΨm(图S4G–S4I)。此外,Bax−/−Bak1−/−Bid−/−细胞中的氧化应激水平明显低于对照细胞(图S4J)。相比之下,NLRP3缺陷不影响线粒体损伤(图S4K–S4M),与Nlrp3−/−细胞对IIAMD诱导的细胞死亡易感一致(图2A)。BAX和BAK1还可通过线粒体转换孔和亲环蛋白D诱导caspase非依赖性细胞死亡,这可被环孢菌素A(CsA)抑制。然而,作者发现CsA处理(图S4N和S4O)或siRNA敲低Ppif(亲环蛋白D编码基因)(图S4P和S4Q)对mitoxyperilysis均无保护效果。总之,这些数据表明BAX、BAK1和BID是响应IIAMD驱动炎症性细胞死亡的线粒体损伤和氧化应激的关键上游调控因子。
7. mTORC2而非mTORC1促进IIAMD下的细胞死亡
IIAMD通过线粒体损伤依赖性细胞死亡mitoxyperilysis诱导氧化应激,其中膜破裂和细胞死亡之前存在损伤线粒体与质膜的长时间接触。为了理解调控mitoxyperilysis的机制,作者筛选了一个包含2,050个小分子、针对约1,000个细胞通路的文库,以鉴定响应LPS加CS的细胞保护化合物(图4A和4B)。除鉴定TLR4抑制剂作为内部验证外,作者发现十种最有效的细胞保护化合物中有三种是mTOR激酶抑制剂(图4B)。RNA-seq鉴定mTOR通路为LPS加CS中显著上调的通路之一(图S5A)。作者还观察到响应mitoxyperilysis触发因素时mTORC1活性(核糖体蛋白S6的S235/236磷酸化)和mTORC2活性(AKT的S473磷酸化)的存在(图S5B)。LPS或PAM3加CS通过MyD88激活mTOR,而TNF加CS通过TNFR信号传导激活mTOR(图S5C)。Bax−/−Bak1−/−Bid−/−细胞的mTOR激活也降低(图S5D)。总之,这些结果提示mTOR在TLR/TNFR信号传导和BAX/BAK1/BID下游对驱动mitoxyperilysis发挥关键作用。
作者接下来确认了筛选中鉴定的三种mTOR抑制剂以剂量依赖方式抑制mitoxyperilysis的能力(图4C)。作者加入了另一种常用的mTOR抑制剂Torin-1,发现它显著降低了细胞死亡、LDH释放以及IL-1β和IL-18释放(图4D和S5E–S5G)。Torin-1对细胞死亡的抑制独立于炎症小体机器(图S5E和S5F),强调炎症小体激活并不驱动这种细胞死亡。Torin-1还抑制LPS加CS诱导的人外周血单核细胞来源的巨噬细胞(huMACs)的细胞死亡(图4E和4F),表明该通路在小鼠和人之间的调控相似。
Torin-1是mTORC1/2双重抑制剂。使用siRNA确定细胞死亡是mTORC1还是mTORC2依赖性的,作者发现Rictor(mTORC2调控因子)而非Rptor(mTORC1调控因子)对LPS加CS诱导的细胞死亡至关重要(图4G和4H)。雷帕霉素抑制mTORC1对LPS加CS诱导的细胞死亡无影响(图S5H和S5I),进一步提示mTORC2而非mTORC1调控mitoxyperilysis。
LPS是公认的mTOR激活剂,提示mTOR抑制剂可能非特异性抑制LPS信号传导从而阻断mitoxyperilysis。然而,Torin-1同样抑制由TNF或其他TLR配体加CS诱导的细胞死亡(图S5J)。此外,Torin-1抑制LPS加CS反应中的mTOR激活,但不影响传统上与LPS信号传导相关的通路激活(图S5K)。这些结果提示mTOR特异性参与调控细胞死亡,而非更广泛地影响LPS信号传导。
总之,这些数据表明mTOR被激活并在功能上诱导mitoxyperilysis是必需的,其中mTORC2特别关键。
8. mTOR激活和氧化应激独立驱动mitoxyperilysis
mTOR抑制可降低线粒体氧化功能并促进自噬,这两者均可降低氧化应激,从而减少mitoxyperilysis。mTOR抑制在未刺激和LPS加CS处理的细胞中降低了p62表达(指示自噬增加)(图S5L)。然而,这种增加的自噬并未伴随细胞氧化应激的降低(图S5M和S5N)。此外,mTOR抑制期间总GSH仍保持低水平(图4I)。一致地,IIAMD中受损的线粒体呼吸作用未被mTOR抑制恢复,尽管可被抗氧化剂NAC处理部分恢复(图4J和4K)。用LPS加CS处理的细胞在mTOR抑制后维持高水平的氧化应激,但长时间保持PI不通透(图4L)。氧化应激的中和不影响mTOR激活(图S5O)。总之,这些数据表明mTOR激活和氧化应激在mitoxyperilysis中是独立事件,mTOR在IIAMD反应中在线粒体功能障碍下游促进细胞死亡。

图4. mTORC2驱动对先天性免疫激活和代谢紊乱的mitoxyperilysis反应
9. mTOR活性通过抑制细胞骨架活性延长线粒体-质膜接触
为了进一步了解mTOR调控mitoxyperiosis的机制,作者进行了活细胞成像。与作者在功能性mTOR的IIAMD处理细胞中观察到的延长线粒体-膜接触相反(图3E和3J;见视频S1、S2和S8),用mTOR抑制剂Torin-1处理的细胞频繁形成片状伪足(图5A;见视频S9)。膜相关线粒体被片状伪足频繁形成而远离质膜,导致接触持续时间缩短(图S6A)并恢复无线粒体的局部质膜(图5A;视频S9)。TEM证实,在mTOR抑制下的IIAMD刺激期间,片状伪足包围了含有线粒体样细胞器的受损质膜(图5B)。使用QuimP测量轮廓位移以计算局部膜区域的移动速度,作者发现IIAMD显著降低了细胞膜动态和片状伪足形成(图5C和5D;见视频S10)。mTOR抑制增加了膜动态(图5C和5D;视频S10),与观察到的频繁片状伪足活性一致(图5A)。
片状伪足活性和细胞膜动态受RhoA和其他控制肌动蛋白聚合的Rho-GTPases调控。确实,RhoA活性在LPS加CS反应中降低,这可被mTOR抑制部分恢复(图5E)。受激发射损耗荧光寿命成像显微镜(tauSTED)提供约100nm分辨率,显示在LPS加CS反应中,外周分布的线粒体(TOM20染色)周围几乎没有F-actin(鬼笔环肽染色),而Torin-1诱导形成具有密集F-actin皱褶的片状伪足以包含突出的线粒体(图5F)。这些观察进一步提示细胞骨架活性和肌动蛋白网络对调控mitoxyperiosis和控制IIAMD期间线粒体-膜接触至关重要。
为了检查细胞骨架活性在mitoxyperiosis和mitoxyperilysis中的功能,作者使用细胞松弛素D(CyD)破坏肌动蛋白纤维(图S6B);CyD消除了Torin-1的作用,显著减少了片状伪足形成和回缩(图5C和5D;视频S10),导致线粒体-膜接触时间延长(图5G和S6C;见视频S11)。CyD对对照条件下的细胞死亡无影响(图5H和S6D),并逆转了Torin-1减少mitoxyperilysis和LDH释放的能力(图S6E)。虽然Rictor敲低保护细胞免于mitoxyperilysis,但CyD阻断了这种保护(图5I)。总之,这些结果提示mTORC2介导的片状伪足形成调控在IIAMD期间控制mitoxyperiosis和mitoxyperilysis中发挥关键作用。
作者接下来使用著名的ARP2/3抑制剂CK-666抑制片状伪足形成。CK-666剂量依赖性地在Torin-1存在下恢复细胞死亡(图S6F和S6G),进一步提示Torin-1通过允许片状伪足形成抑制细胞死亡。相反,微管破坏剂诺考达唑(图S6B)不影响细胞死亡(图S6H)。总之,这些数据表明,在IIAMD反应中,mTOR抑制肌动蛋白聚合相关的细胞骨架活性,以允许延长线粒体-膜接触并驱动mitoxyperilysis。mTOR抑制增强细胞膜动态和片状伪足活性,使线粒体远离质膜并保护膜完整性。
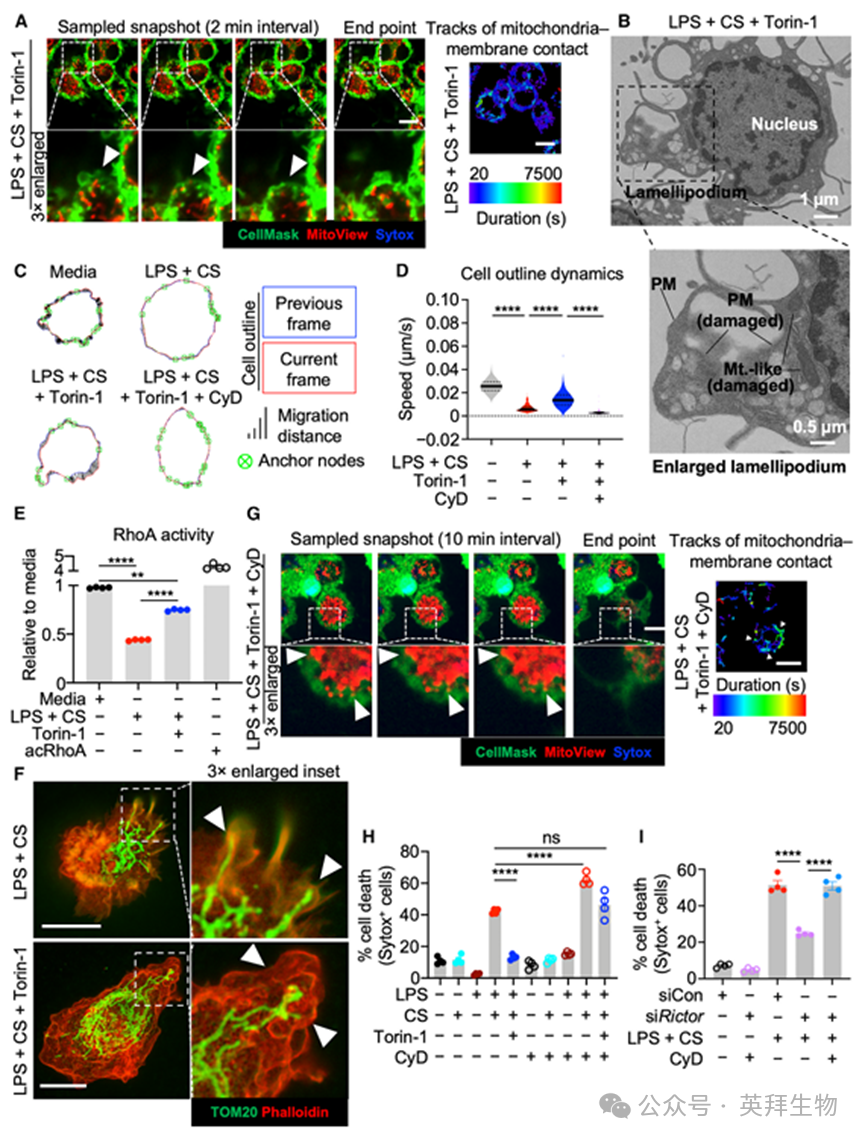
图5. mTOR通过抑制细胞骨架活性促进线粒体在质膜的持续滞留以驱动mitoxyperilysis
10. 响应IIAMD的细胞死亡的机制特征定义了一种独特的细胞死亡通路
鉴于线粒体功能障碍、细胞骨架活性、氧化应激和mTORC2在mitoxyperilysis中的显著参与,作者试图更好地表征其定义性特征,并与其它细胞死亡通路进行比较。作者选择了由各自经典触发因素诱导的凋亡、NLRP3炎症小体诱导的细胞死亡、坏死性凋亡和铁死亡(分别为星形孢菌素、LPS加尼日利亚菌素、TNF加SMAC模拟物和z-VAD [TSZ]、以及RSL3)进行比较。作者首先评估了线粒体功能障碍动力学。LPS加CS随时间点降低了ΔΨm(图6A),而在凋亡中ΔΨm最初升高然后逐渐降低(图6B)。在NLRP3炎症小体诱导的细胞死亡、坏死性凋亡和铁死亡中观察到持续升高的ΔΨm(图6C–6E)。作者还评估了细胞骨架动态,发现RhoA活性仅在LPS加CS反应中独特降低,而在其它细胞死亡方式中未降低(图6F–6J)。在所有五种细胞死亡方式中均观察到氧化应激,其中LPS加CS和铁死亡最显著地降低GSH(图6K)。mTOR在凋亡或NLRP3炎症小体诱导的细胞死亡中无调控作用(图6L和6M)。Torin-1和雷帕霉素均促进坏死性凋亡(图6N)并减少铁死亡(图6O),提示在这些情况下mTORC1起主要作用,与LPS加CS诱导的细胞死亡中mTORC2的主要作用形成对比(图4H和S5H)。
总之,这些比较评估进一步凸显了mitoxyperilysis背后的独特机制特征,这将其与其它已建立的细胞死亡方式区分开来。

图6. 先天性免疫激活和代谢紊乱诱导的细胞死亡具有与其他细胞死亡通路不同的独特特征
11. IIAMD以mTOR依赖性方式驱动炎症性疾病死亡率
作者之前观察到,与单独LPS挑战相比,LPS挑战前禁食导致更高的死亡率和组织损伤(图S1A–S1C)。鉴于在体外鉴定了IIAMD诱导的细胞死亡通路mitoxyperilysis,作者接下来评估其在体内的相关性(图S7A)。与mTOR在体外驱动mitoxyperilysis的作用一致,mTOR抑制显著降低了禁食小鼠LPS休克中的死亡率,并降低了组织损伤和肝功能障碍标志物(图S7B–S7D)。在进食条件下,LPS诱导的死亡率由GSDMD介导。Gsdmd−/−小鼠对无禁食的LPS挑战有抵抗力;然而,Gsdmd−/−小鼠预先禁食导致显著的LPS休克死亡率(图S7E)。这些发现表明IIAMD触发mTOR介导的细胞死亡,导致体内组织损伤和死亡率,且独立于传统的GSDMD依赖性通路。
12. IIAMD在体外和体内以mTOR依赖性方式诱导肿瘤细胞死亡
为了确定mitoxyperilysis的潜在治疗意义,作者聚焦于已知对免疫和代谢操作均敏感的肿瘤微环境。小鼠B16黑色素瘤细胞对TLR配体有反应(图S7F),并对单独CS发生中度细胞死亡(图S7G);然而,IIAMD显著增加了细胞死亡(图S7G)。与原代巨噬细胞相比,B16细胞具有更高的基础氧化应激和mTOR信号水平(图S7H和S7I),这可能促成其对CS单独处理的细胞死亡易感性。确实,抑制mTOR信号传导显著降低了所有测试条件下的细胞死亡,包括单独CS(图S7G)。
基于作者观察到的MyD88和TNFR在介导BMDMs中IIAMD诱导的细胞死亡中的关键作用(图1J和1K),作者试图确认这种先天性免疫信号传导在癌细胞死亡中的作用。与作者在巨噬细胞中的发现一致,LPS加CS诱导的B16细胞死亡被Myd88缺陷减少,但不被Tnfrsf1a缺陷减少(图S7J和S7K),而TNF加CS诱导的细胞死亡依赖于TNFR并绕过对MyD88的需求(图S7J和S7K)。为了进一步确认mTOR在癌细胞死亡中的作用,作者使用CRISPR生成了Rptor(mTORC1缺陷)和Rictor(mTORC2缺陷)敲除B16肿瘤系(图S7L)。Rictor−/−而非Rptor−/− B16细胞对LPS加CS和TNF加CS反应的细胞死亡降低(图S7M),提示在细胞类型间mitoxyperilysis中存在保守的mTORC2依赖性机制。
作者接下来试图在体内应用这种治疗策略。在B16黑色素瘤异位肿瘤模型中,荷瘤小鼠接受低剂量LPS的瘤内(i.t.)注射并结合禁食(图7A)。该治疗破坏了能量代谢,因为肿瘤间质液中的葡萄糖、许多TCA循环相关代谢物和氨基酸水平显著降低(图7B)。LPS i.t.加禁食后,肿瘤大小显著减小(图7C),坏死增加(图S7D和S7E);相比之下,单独禁食或LPS i.t.治疗通常仅阻止肿瘤生长但未减小肿瘤大小(图7C)。单独禁食小鼠是肿瘤抑制而非肿瘤杀伤性的,因为它与LPS i.t.加禁食相比未诱导显著坏死(图S7N)。
作者接下来检查了mTOR信号传导在这种肿瘤消退中的作用。mTORC1信号传导在LPS i.t.加禁食反应中降低(图S7O),可能由于氨基酸水平降低。此外,mTORC2信号传导增强(图S7O),与先前禁食条件下的观察一致。作者接下来在禁食和LPS i.t.前用Torin-1治疗荷瘤小鼠。mTOR抑制降低了联合治疗效果,导致肿瘤大小无显著变化且肿瘤坏死减少(图7C–7E)。作者还将Rptor−/−、Rictor−/−和对照B16肿瘤细胞植入小鼠并用禁食和LPS i.t.治疗。虽然Rptor−/−肿瘤大小减小且这些肿瘤的坏死与对照肿瘤相当,但Rictor−/−肿瘤对治疗的抵抗部分丧失,无显著大小减小且坏死较少(图7F和7G)。这些结果提示,肿瘤固有mTORC2功能对禁食和LPS i.t.治疗诱导的体内细胞死亡至关重要,与作者体外结果一致。
结合对其它关键细胞死亡特征的比较,这些结果显示响应先天性免疫激活和代谢紊乱的细胞死亡与先前充分表征的通路不同。
此外,IIAMD要素之间的协同作用对驱动这种细胞死亡至关重要。单独的先天性免疫激活(如LPS刺激)诱导氧化应激和mTOR激活,但也增加GSH水平以对抗氧化应激并阻止死亡。同样,单独的代谢紊乱触发一些线粒体缺陷,但未诱导强烈的氧化应激和mTOR信号传导,阻止了溶解性细胞死亡。这些组分的协同作用是实现驱动强烈细胞死亡所需的线粒体功能障碍、氧化应激和mTOR激活水平所必需的。
为了进一步确认体内肿瘤缩小与mitoxyperiosis相关,作者测量了肿瘤裂解物中的RhoA活性。与LPS i.t.加禁食治疗24h后,肿瘤中RhoA活性显著降低,而添加Torin-1治疗可部分恢复RhoA活性(图S7P),与体外结果一致(图5E和6F)。坏死肿瘤在质膜附近含有损伤的线粒体样细胞器(图7H),支持膜邻近线粒体损伤在肿瘤细胞死亡中的作用。这些数据表明,mitoxyperilysis可被治疗性激活以减少肿瘤负荷。

图7. 先天性免疫激活和代谢紊乱的协同作用以mTORC2依赖性方式诱导肿瘤坏死和缩小
参考文献
1、Wang Y, Lu J, Carisey AF, Chadchan SB, Lee HW, Malireddi RKS, Sharma BR, Pandian N, Tweedell RE, Palacios G, Becerra Mora N, Robinson CG, Pitre A, Vogel P, Chen T, Murphy MP, Kanneganti TD. Innate immune and metabolic signals induce mitochondria-dependent membrane lysis via mitoxyperiosis. Cell. 2025 Nov 28:S0092-8674(25)01251-6.

本周
本月
本年
LetPub发布最新SCI影响因子查询及期刊投稿分析系统
草甘膦(glyphosate)酶联免疫分析(ELISA)试剂盒使用说明书
ELISpot试剂盒限时特惠!一口价低至500元,加赠免费读板服务
单个细胞也能提取核酸?超全干货教你微量样本发高分(含完整电子版宝典资料))
应用gentleMACS™灌流技术从脂肪肝小鼠模型高效分离肝细胞与非实质细胞
LetPub完整SCI影响因子、期刊分区查询系统
Elabscience® 从原料到标记,打造属于中国自己的流式抗体品牌
链脲佐菌素 (Streptozotocin,STZ)-糖尿病动物模型造模
11月Cell重磅:新线粒体细胞死亡方式!或将引爆国自然新热点!先天免疫和代谢信号通过线粒体周膜裂解诱导线粒体依赖性膜裂解
【阿拉丁】免疫荧光实验方案
- 促销公告
- 更多 ›
