

葡萄糖富集的肺转移前微环境由肝细胞癌中基质刚度调控的外泌体miRNA触发
2025-09-22 14:36点击次数:215
关键词:除经典特征外,肝细胞癌(HCC)转移前微环境形成过程中是否存在其他新型病理性特征尚属未知。我们前期的研究揭示了基质硬度增加对HCC肺转移前微环境形成及转移的促进作用。然而,基质硬度增加是否会影响肺转移前微环境的葡萄糖代谢与供给仍不明确。本研究揭示了基质刚度调控的外泌体miRNA通过降低肺成纤维细胞的葡萄糖摄取与消耗、同时增强血管生成和血管通透性,从而调节葡萄糖在肺转移前微环境富集的作用机制。我们的研究结果表明,由基质刚度调控的外泌体miRNA触发的葡萄糖富集现象,是肺转移前微环境的新特征,对转移性肿瘤细胞的定植、存活以及后续转移灶的生长至关重要。该研究于2025年2月发表在《Nature Communication》,IF:15.7。
技术路线:
主要研究结果:
1.在高硬度基质上培养的HCC细胞来源的条件培养基可加速肺转移前微环境的形成
参考本课题组先前报道的转移前微环境动物模型诱导方法37,我们建立了另一种无肿瘤肺转移前微环境小鼠模型,以评估高硬度刺激下HCC细胞条件培养基对肺转移前微环境形成的贡献。如补充图1a和图1a动物实验示意图所示,我们分别收集了在6 kPa和16 kPa基质上培养的Hepa1-6细胞条件培养基(分别命名为L-CM和H-CM),通过尾静脉隔日注射至小鼠(C57BL/6)体内,诱导无肿瘤肺转移前微环境模型。注射的L-CM和H-CM分别代表具有正常肝背景和肝硬化背景的HCC肿瘤释放的可溶性因子。考虑到骨髓源性细胞(BMDCs)招募是肺转移前微环境最显著的特征之一2,我们通过流式细胞术检测了不同诱导时间点新鲜肺组织中CD11b+CD45+ BMDCs的招募水平。随着诱导天数增加,L-CM组和H-CM组的CD11b+CD45+ BMDCs数量均有所增加。同时,与L-CM组相比,H-CM组在第18、22和26天的CD11b+CD45+ BMDCs招募水平均显示增强,其中第26天BMDCs招募差异最为显著(图1b,c)。上述结果表明H-CM可能具有较强的诱导HCC肺转移前微环境形成的能力。此外,这些结果使我们初步确定第26天为无肿瘤肺转移前微环境小鼠模型成功建立的诱导时间点。我们进一步分析了第26天这些动物模型新鲜肺组织中转移前微环境相关基因(Fn、Mmp9、S100a8、S100a9和Bv8)的表达,发现H-CM组转移前微环境相关基因的表达水平均显著高于L-CM组(图1d),与该组BMDCs招募结果一致。相应地,我们还观察到H-CM组纤维连接蛋白(FN)表达(图1e)和髓源性抑制细胞(MDSCs)比例明显增加(图1f),以及效应CD8+ T细胞比例显著降低(图1g),证实H-CM确实促进肺转移前微环境形成。此外,我们检测了第26天肺组织血管密度和通透性的分子标志物,结果显示H-CM组CD31表达明显增加,而CD31+区域内VE-钙黏蛋白覆盖度显著降低(图1h,i),表明H-CM具有更强的促进血管生成和破坏血管完整性的能力。基于两组在BMDCs招募、基质重塑(FN和MMP9)、免疫抑制(MDSCs和CD8+ T细胞)、血管生成与血管通透性(BV8、CD31和VE-钙黏蛋白)以及炎症(S100A8和S100A9)方面的显著差异,我们得出结论:与L-CM相比,H-CM显著加速了肺转移前微环境的形成。
除上述体内实验结果外,我们随后参照既往方法37构建了模拟病理状态下肺组织硬度(926.18 Pa,肺硬度基质)的体外凝胶基质培养定居细胞,并采用H-CM干预这些细胞以模拟体内肺转移前微环境。我们分别使用在6 kPa和16 kPa硬度基质上培养的人HCC细胞来源的L-CM和H-CM处理肺硬度基质上生长的肺成纤维细胞,发现与L-CM干预相比,H-CM干预显著上调了肺成纤维细胞中FN和MMP9的蛋白水平(图1j),与动物实验结果一致(图1d)。qPCR和ELISA分析也显示H-CM干预明显促进肺成纤维细胞中FN和MMP9的基因和蛋白表达(补充图1b,c)。同时我们观察到,经H-CM处理的肺成纤维细胞单层表面黏附的HCC细胞数量显著增加(图1k)。这些结果强有力地证明H-CM通过影响肺成纤维细胞促进肺基质重塑,为肿瘤细胞黏附创造适宜环境。另一方面,我们评估了H-CM对血管内皮细胞(HUVECs)中VEGFR2、ZO-1和VE-钙黏蛋白表达的影响,结果显示H-CM显著提高VEGFR2表达,同时抑制ZO-1和VE-钙黏蛋白表达(图1l),表明H-CM具有更强的促进血管生成和破坏血管完整性的诱导能力,这与动物模型中的发现一致(图1h,i)。此外,经H-CM处理的HUVEC单层对FITC-葡聚糖的通透性也出现增加(图1m),证实H-CM可通过调控血管内皮细胞显著增加血管通透性。综上,体内外证据充分表明,高硬度刺激下HCC细胞释放的条件培养基能显著促进肺转移前微环境的形成。
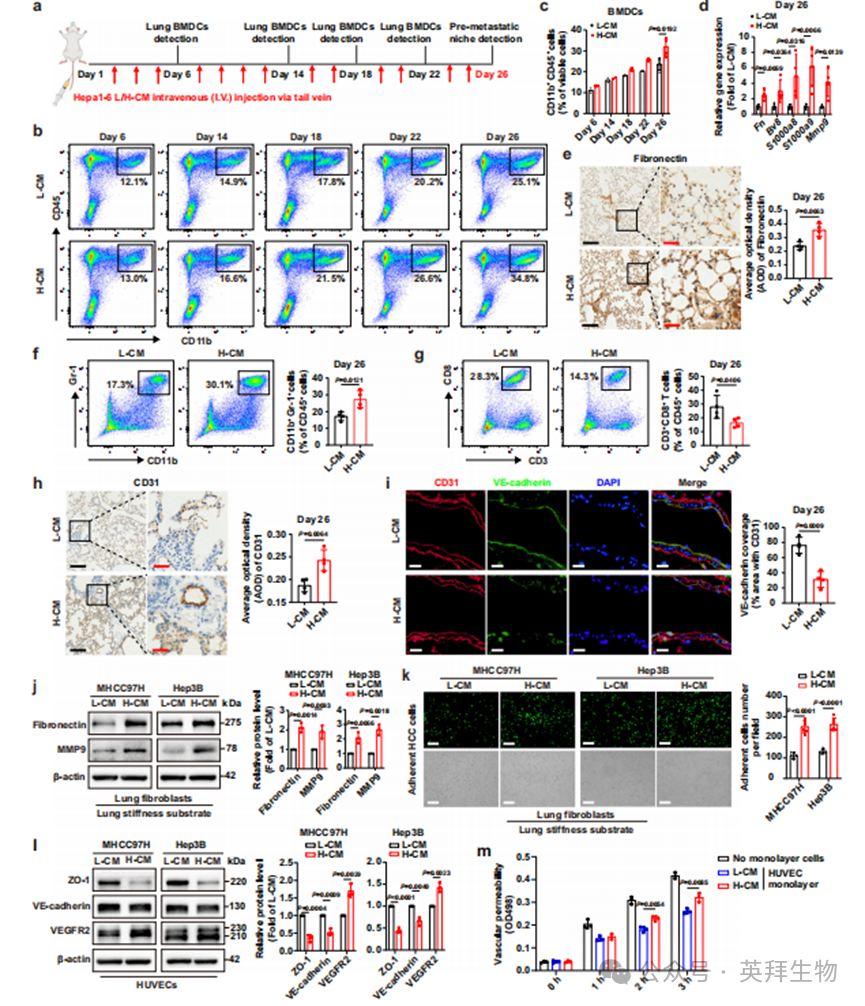
图1.在高硬度基质上培养的HCC细胞来源的条件培养基可加速肺转移前微环境的形成。
2.葡萄糖富集现象发生于H-CM诱导的肺转移前微环境形成过程中
转移靶器官中的定居细胞(成纤维细胞、血管内皮细胞等)在肿瘤源性可溶性因子的调控下,通常参与重塑利于循环肿瘤细胞定植与存活的"土壤"环境4-8。我们前期的研究也表明,高硬度刺激下HCC细胞分泌的LOXL2可通过调控肺成纤维细胞和招募BMDCs促进肺转移前微环境形成37,38。葡萄糖作为主要营养物质,主要满足肿瘤细胞增殖与存活的能量需求,而重编程的葡萄糖代谢也被视为癌症的典型特征18。肺转移前微环境是HCC细胞的异位存活场所,其中葡萄糖富集可能对支持播散肿瘤细胞的异位定植、存活和生长至关重要。因此,除BMDCs招募、基质重塑、免疫抑制、炎症反应、血管生成和血管通透性等经典病理改变外,我们推测葡萄糖代谢重编程也可能发生于转移前微环境形成过程中。为探究高硬度刺激下HCC细胞释放的条件培养基是否调控肺转移前微环境中定居细胞的葡萄糖代谢,我们采用上述模拟肺转移前微环境的实验体系,评估基质细胞中葡萄糖转运蛋白和糖酵解酶的表达变化。结果显示,H-CM干预显著抑制肺成纤维细胞中GLUT1、PFKP、PKM2和HK2的表达,但对SGLT2影响甚微(图2a),这意味着肺成纤维细胞的葡萄糖摄取与代谢水平均严重减弱。一致性地,2-NBDG摄取实验和葡萄糖消耗assay 也显示H-CM处理的肺成纤维细胞葡萄糖摄取与消耗明显降低(图2b,c)。随后我们通过非靶向代谢组学分析进一步明确H-CM对肺成纤维细胞代谢变化的影响,发现H-CM干预导致细胞内糖酵解代谢物(包括6-磷酸葡萄糖、6-磷酸果糖、2-磷酸-D-甘油酸和磷酸烯醇丙酮酸)含量显著降低,这些差异代谢物在中心碳代谢通路中富集(图2d和补充图1d)。以上数据均支持H-CM干预有效减弱肺成纤维细胞的葡萄糖摄取与消耗,从而创造葡萄糖富集的微环境。为验证这一发现,我们继续检测无肿瘤肺转移前微环境小鼠模型新鲜肺组织中葡萄糖转运蛋白和糖酵解酶的表达变化。结果同样显示,除Slc5a2(SGLT2编码基因)外,糖酵解酶(Pfkp、Pkm和Hk2)和葡萄糖转运蛋白(Slc2a1)的表达水平在H-CM组均显著降低(图2e)。同时,肺组织水平GLUT1低表达和糖原含量增加(图2f和补充图1e)也支持H-CM组葡萄糖代谢水平显著下降和葡萄糖累积增加。此外,通过调控血管内皮细胞实现的血管通透性和血管生成增强(图1l,m),也提示肺转移前微环境中葡萄糖供给与聚集增加。所有这些数据表明,H-CM诱导的肺转移前微环境形成过程中存在葡萄糖富集这一新特征。为明确葡萄糖富集作为肺转移前微环境新特征的病理学意义,我们开展了系列研究。基于葡萄糖富集有助于增强HCC细胞黏附能力(图2g),且MDSCs聚集可能部分源于其分化增加(图1f)的结果,我们合理推测转移前微环境中的葡萄糖富集可能通过影响黏附分子表达促进HCC细胞黏附,或增强免疫抑制强度以利于肿瘤细胞存活与转移。我们聚焦于能与基质细胞产生的纤连蛋白结合的黏附分子整合素β140,探究高葡萄糖上调整合素β1表达的潜在机制。结果显示高葡萄糖条件显著提升HCC细胞中整合素β1表达水平(补充图2a),表明高葡萄糖与整合素β1表达存在关联。AMPK作为细胞能量感应关键调节因子,既往研究报道其在癌细胞、成肌细胞和足细胞中受高葡萄糖负向调控41-43。鉴于AMPKα可降解SNX17(一种参与整合素β1再循环至质膜的内体循环调节因子44-46),我们进一步推测高葡萄糖可能通过调控AMPKα/SNX17信号通路上调整合素β1。分析HCC细胞在高葡萄糖条件下AMPKα和SNX17的活性与表达,发现高葡萄糖抑制AMPKα磷酸化但提升SNX17表达(补充图2a)。此外,AMPK激动剂A-769662显著逆转高葡萄糖对SNX17和整合素β1的上调作用(补充图2a)。通过膜蛋白提取和免疫荧光染色分析HCC细胞表面整合素β1水平,发现高葡萄糖显著上调其细胞表面表达,而AMPK激活可逆转此效应(补充图2a,b),表明高葡萄糖刺激下整合素β1再循环至质膜增加。为明确高葡萄糖诱导的整合素β1上调是否参与HCC细胞对肺转移前微环境的黏附,我们使用AMPK激动剂(A-769662)或整合素β1功能阻断抗体处理HCC细胞观察黏附变化。结果显示激活AMPK或阻断整合素β1均可显著抑制HCC细胞在肺成纤维细胞单层上的黏附(图2h和补充图2c)。由此可见,肺转移前微环境中的葡萄糖富集可通过调控AMPKα/SNX17通路上调HCC细胞表面整合素β1水平,从而促进肿瘤细胞对转移前微环境的黏附。
除细胞黏附特性外,肺转移前微环境中MDSCs的数量对循环肿瘤细胞在异质组织中的定植与存活也至关重要47。在无肿瘤肺转移前微环境小鼠模型中,我们验证了H-CM对MDSCs在肺转移前微环境中聚集的重要作用(图1f)。考虑到MDSCs聚集可能部分源于其分化增加,我们继续探究葡萄糖富集是否影响MDSCs分化。在IL-6和GM-CSF存在条件下,高葡萄糖显著促进骨髓细胞(BMCs)分化为MDSCs,而糖酵解抑制剂(2-DG)干预显著减弱此分化效应(图2i)。既往研究报道mTOR通路激活可能参与葡萄糖累积对MDSCs分化与功能的影响48,49。本研究中我们同样观察到高葡萄糖刺激下MDSCs中mTOR磷酸化水平升高,但2-DG干预有效抑制此效应(图2j)。类似地,mTOR活性抑制剂雷帕霉素也显著降低高葡萄糖刺激下的MDSCs分化(补充图2d,e),证实葡萄糖富集对MDSCs分化的促进作用。综上,转移前微环境中的葡萄糖富集通过AMPKα/SNX17通路上调整合素β1表达促进HCC细胞对微环境的黏附,同时通过增加MDSCs分化强化免疫抑制微环境以利于后续定植与存活,这些发现凸显葡萄糖富集是肺转移前微环境的一个重要新特征。
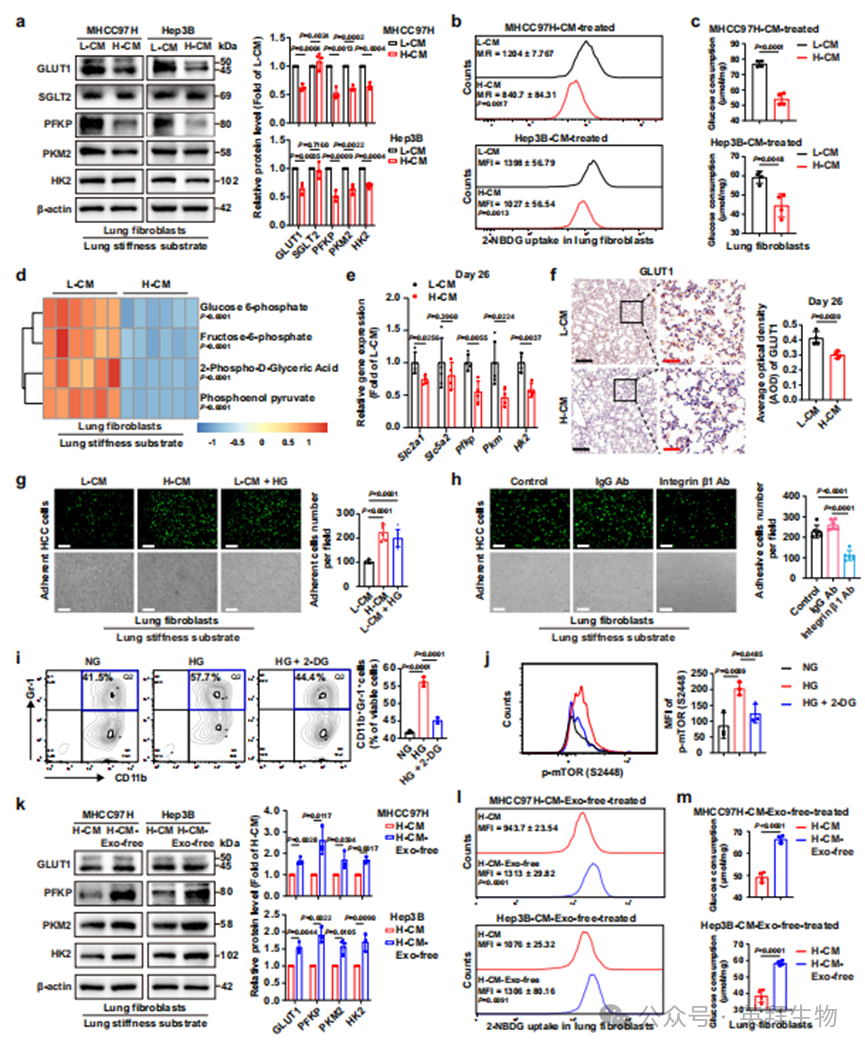
图2.葡萄糖富集现象发生于H-CM诱导的肺转移前微环境形成过程中
3.肿瘤源性外泌体作为主要调控因子介导H-CM诱导的肺转移前微环境中葡萄糖富集
原发性肿瘤释放的可溶性因子通常包含分泌蛋白、细胞外囊泡(EVs)及其他分子元件2,9。外泌体作为分泌型微囊泡可从肿瘤细胞转移至受体细胞实现细胞间信息交流9。考虑到葡萄糖代谢重编程发生于细胞内部,肿瘤细胞来源的外泌体很可能在肺转移前微环境形成过程中主导葡萄糖富集的调控。我们通过差速超速离心从H-CM中制备去外泌体H-CM(H-CM-Exo-free),并通过分析TSG101、CD63、Hsp70和ALIX等外泌体标志物确认其质量(补充图2f)。分别用H-CM-Exo-free和H-CM处理肺硬度基质上生长的肺成纤维细胞,结果显示H-CM-Exo-free干预显著逆转了H-CM对糖酵解酶和葡萄糖转运蛋白的调控作用(图2k),以及葡萄糖摄取与消耗能力(图2l,m)。这些数据表明外泌体确实是H-CM诱导肺转移前微环境过程中调控葡萄糖富集的主要贡献者。
为进一步验证外泌体在H-CM诱导肺转移前微环境中调控葡萄糖富集的主导作用,我们采用差速超速离心法从L-CM和H-CM中纯化外泌体,分别命名为L-Exo和H-Exo。纯化的外泌体呈现典型杯状结构(图3a),平均尺寸介于100-150 nm之间(补充图2g),符合外泌体的典型表型特征。此外,检测到TSG101、CD63、Hsp70和ALIX等分子标志物表达,而白蛋白和细胞色素C等阴性标志物缺失(图3b),进一步证实纯化外泌体质量符合特征要求,可用于下游实验。我们采用膜绿色荧光探针(DIO)标记L-Exo和H-Exo以确定受体细胞,发现与DIO标记外泌体共孵育后,两类受体细胞(肺成纤维细胞和血管内皮细胞)内均出现绿色荧光点(图3c和补充图2h),表明HCC细胞来源的L-Exo和H-Exo可成功转移至肺成纤维细胞和血管内皮细胞。
鉴于肿瘤细胞分泌的外泌体可动员BMDCs至次级器官部位触发转移前微环境形成1,2,我们利用Millicell悬吊式细胞培养插板构建体外共培养系统(图3d)模拟转移前微环境,以阐明外泌体在其中的作用。具体而言,在含IL-6和GM-CSF的下室中培养BMCs 48小时,然后与上室中经L-Exo或H-Exo处理的肺成纤维细胞(培养于肺硬度基质)共培养48小时(补充图2i)。观察到与L-Exo干预相比,H-Exo干预显著提高肺成纤维细胞中FN和MMP9表达,而与BMCs共培养进一步强化这些效应(图3d)。同时,当BMCs与H-Exo处理的肺成纤维细胞共培养时,其向MDSCs分化的比例显著增加(图3e)。H-Exo干预结果与体内H-CM干预效果基本一致,表明H-Exo与H-Exo类似,也能促进肺转移前微环境形成。
采用L-Exo或H-Exo处理肺硬度基质上的肺成纤维细胞,我们进一步检测糖酵解相关蛋白表达和葡萄糖摄取能力以验证其对葡萄糖代谢的调控作用。结果显示H-Exo干预显著降低肺成纤维细胞中葡萄糖转运蛋白(GLUT1)和糖酵解酶(PFKP、PKM2和HK2)的基因与蛋白表达水平(补充图3a-c)。此外,H-Exo处理也减弱了其葡萄糖摄取与消耗能力(补充图3d,e),与H-CM干预结果一致(图2a-c),证实外泌体对葡萄糖代谢的调控作用。同时发现经H-Exo孵育的肺成纤维细胞单层表面黏附的HCC细胞数量增加(补充图3f),表明H-Exo通过重塑基质和促进葡萄糖富集,为CTCs黏附与定植创造有利的"土壤"环境。
另外,与L-Exo处理的对照组细胞相比,H-Exo处理的HUVECs显示更高VEGFR2表达和更低ZO-1与VE-钙黏蛋白表达(补充图3g)。此外,H-Exo处理促进HUVECs成管能力(补充图3h)并增强HUVEC单层对FITC-葡聚糖的通透性(补充图3i),提示H-Exo干预通过改变血管通透性和血管生成增加肺转移前微环境中的葡萄糖供给与聚集。
为在体内进一步验证H-Exo对肺转移前微环境形成(尤其是葡萄糖富集)的贡献,我们建立了H-Exo诱导的无肿瘤肺转移前微环境小鼠模型。从Hepa1-6-L-CM或H-CM中分离外泌体后,通过外泌体分子标志物分析确认其质量(补充图3j)。参照图1a所述方法,我们通过尾静脉隔日分别向C57BL/6小鼠体内注射L-Exo和H-Exo以诱导肺转移前微环境形成(图3f)。在外泌体诱导第26天,收集新鲜肺组织进行后续分析。与H-CM诱导结果一致,H-Exo诱导同样显著促进肺转移前微环境形成,包括增加CD11b+CD45+ BMDCs招募、诱导基质重塑、促进血管生成和肺部炎症反应(图3g和补充图3k)。同时,H-Exo诱导也抑制了肺组织中糖酵解酶(Pfkp、Pkm和Hk2)和葡萄糖转运蛋白(Slc2a1)的表达水平(补充图3l)。
我们从小鼠肺组织分离收集间质液,检测无细胞间质液中的葡萄糖水平。结果显示H-Exo诱导组小鼠肺组织间质葡萄糖含量高于L-Exo诱导组(图3h),表明H-Exo诱导的肺转移前微环境中存在葡萄糖富集。此外,对外泌体标志物TSG101、成纤维细胞标志物S100A4(FSP-1)和葡萄糖转运蛋白GLUT1进行多重免疫荧光分析显示,在H-Exo诱导组的外泌体富集区域,肺成纤维细胞成功摄取外泌体且其葡萄糖摄取能力(GLUT1表达)明显降低(补充图3m)。进一步地,H-Exo诱导还通过抑制CD31阳性血管内皮细胞中VE-钙黏蛋白表达来增加血管通透性(图3i),提示H-Exo可增加葡萄糖供给与聚集。
值得注意的是,H-Exo诱导还促进MDSCs在转移前肺组织中的聚集,并伴随mTOR活性增强(补充图3n),同时降低CD8+ T细胞浸润(补充图3o),进一步证实葡萄糖富集在强化肺转移前微环境免疫抑制中的作用。我们继续向已建立的动物模型静脉注射Hepa1-6-Luc细胞(图3f),发现H-Exo诱导的葡萄糖富集型肺转移前微环境显著促进HCC细胞体内定植和肺转移(图3j,k),验证了葡萄糖富集型肺转移前微环境对转移性肿瘤细胞定植、存活及后续转移灶生长的促进作用。综上,这些发现表明外泌体不仅介导基质硬度诱导的肺转移前微环境形成,更是导致肺转移前微环境形成过程中葡萄糖富集的主要贡献者。
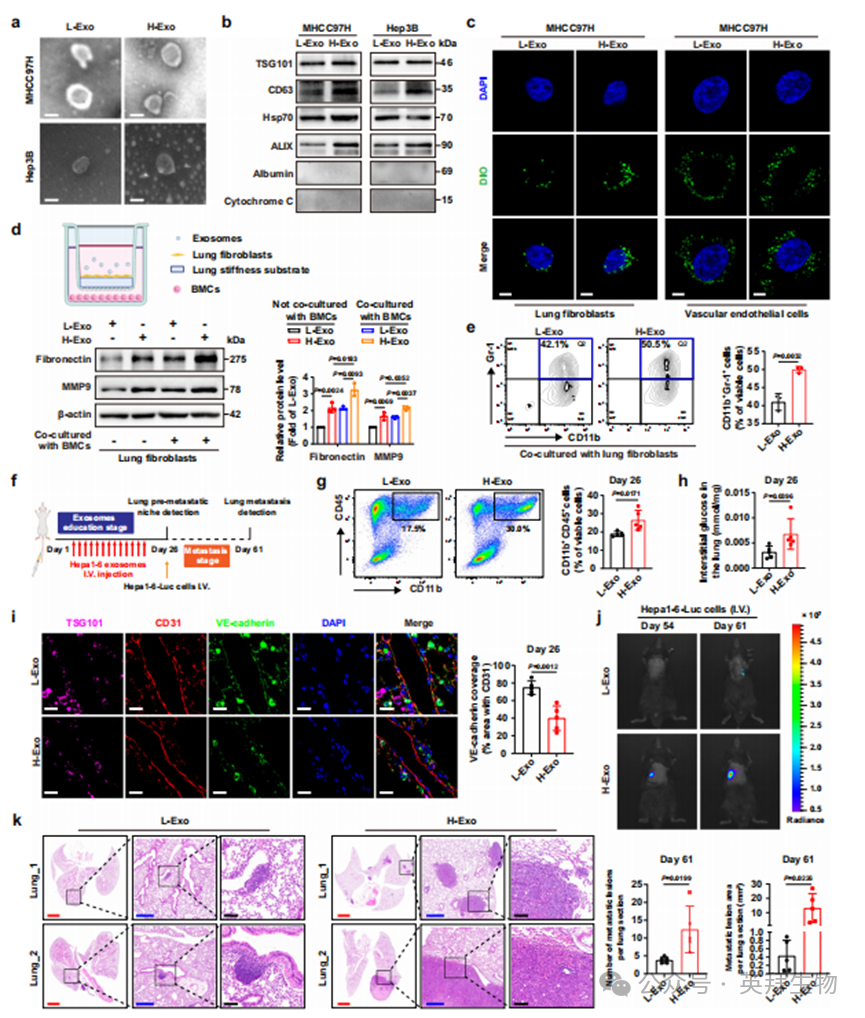
图3.肿瘤源性外泌体作为主要调控因子介导H-CM诱导的肺转移前微环境中葡萄糖富集
4.基质刚度调控的外泌体let-7d-5p促进肺转移前微环境形成过程中的葡萄糖富集
由于miRNA在基因表达调控中的高效性50,外泌体中的miRNA常被视为细胞间通讯的重要介质。我们进一步探究基质刚度调控的外泌体中哪些miRNA参与肺转移前微环境形成过程中的葡萄糖富集。通过小RNA测序筛选MHCC97H-L-Exo与MHCC97H-H-Exo之间的差异表达miRNA。以|FC|>1.5(FC:倍数变化)且P<0.05为阈值,在MHCC97-H-Exo中共鉴定出15个已知差异表达miRNA(包括9个上调和6个下调miRNA)(图4a),以及15个新型未知差异表达miRNA(补充图4a)。采用miRNA qRT-PCR随机验证部分差异miRNA(let-7d-5p、miR-365a-5p、miR-194-5p、miR-125a-5p)的表达,这些miRNA在MHCC97H-H-Exo中均显著上调(图4b),表明测序结果可靠。而在Hep3B-H-Exo中,除miR-194-5p外,这些miRNA同样呈现过表达(图4b)。此外,我们在细胞内表达水平也观察到这些miRNA的相似表达趋势(补充图4b),表明这些miRNA在细胞外泌体与细胞内的表达趋势存在一致性。
随后我们分别将9个上调miRNA的模拟物转染至肺成纤维细胞,以评估不同miRNA对葡萄糖富集的贡献。糖酵解相关基因分析表明,与其他miRNA模拟物相比,let-7d-5p模拟物对SLC2A1、PFKP、PKM和HK2表达的抑制作用最为显著(图4c),提示H-Exo中的let-7d-5p可能是调控肺成纤维细胞葡萄糖代谢的关键miRNA。另一方面,我们采用慢病毒介导的过表达或RNAi技术构建let-7d-5p过表达或敲低的肺成纤维细胞(补充图4c),并在模拟肺转移前微环境的肺硬度基质上培养,分析糖酵解相关蛋白表达。结果显示let-7d-5p过表达显著抑制GLUT1、PFKP和HK2的表达,而let-7d-5p敲低则明显提升其表达(图4d)。有趣的是,在let-7d-5p过表达的肺成纤维细胞中,我们观察到PKM在mRNA水平下降(图4c),但蛋白水平变化不明显(图4d)。此外,我们还评估了let-7d-5p对葡萄糖摄取的影响,发现let-7d-5p过表达明显降低肺成纤维细胞的葡萄糖摄取,而let-7d-5p敲低则提升其摄取能力(图4e),验证了let-7d-5p上调对肺转移前微环境形成过程中葡萄糖富集的促进作用。
为了验证外泌体let-7d-5p在体内促进葡萄糖富集的作用,我们通过尾静脉注射let-7d-5p-OE/Exo,建立了无肿瘤小鼠模型,使其形成肺部前转移灶。我们利用慢病毒介导的过表达技术构建了let-7d-5p过表达的Hepa1-6细胞(Hepa1-6-let-7d-5p-OE)(补充图4d),并收集其培养上清以提取外泌体。随后,我们确定了纯化外泌体的质量符合外泌体的特征(补充图4e、f)。更重要的是,与对照组相比,来自Hepa1-6-let-7d-5p-OE的细胞外外泌体中let-7d-5p的表达水平显著增强(补充图4d),这表明let-7d-5p-OE/Exo有资格作为诱导前转移灶动物模型的介质。与图1a和图3f中描述的方法类似,我们每隔一天通过尾静脉将let-7d-5p-OE/Exo或Mock/Exo注射到C57BL/6小鼠体内,以诱导肺部前转移灶的形成。我们在外泌体诱导的第26天观察到,在let-7d-5p-OE/Exo诱导的小鼠新鲜肺组织中,CD11b+CD45+骨髓来源的树突状细胞(BMDCs)的比例显著增加(图4f),这突出了外泌体let-7d-5p在促进肺部前转移灶形成中的重要作用。此外,我们还检测了外泌体诱导的肺部前转移灶动物模型中肺成纤维细胞(CD45-CD31-CD140a+)的葡萄糖代谢,发现与Mock/Exo组相比,let-7d-5p-OE/Exo组的肺成纤维细胞对2-NBDG的吸收显著减少(图4g)。此外,let-7d-5p-OE/Exo处理显著抑制了肺组织中GLUT1、PFKP和HK2的表达(图4h),并增加了糖原含量(补充图4g),这证实了外泌体let-7d-5p在肺部前转移灶形成过程中葡萄糖富集的显著作用。
我们继续测量外泌体诱导的肺部前转移灶动物模型中新鲜肺组织中MDSCs和CD8+ T细胞的比例。我们观察到let-7d-5p-OE/Exo组中MDSCs的比例明显增加,而CD8+ T细胞的比例显著减少(补充图4h、i),这间接反映了肺部前转移灶中MDSCs的积累部分归因于葡萄糖富集,与体外发现高葡萄糖促进MDSCs分化的结果一致(图2i)。因此,葡萄糖富集可能参与加强肺部前转移灶中的免疫抑制微环境。为了明确let-7d-5p-OE/Exo诱导的肺部前转移灶动物模型对肝细胞癌(HCC)转移的影响,我们将HCC细胞通过尾静脉注射到肺部前转移灶动物模型中,并在第61天(HCC细胞注射后35天)检测两组小鼠肺转移的发生情况。结果表明,let-7d-5p-OE/Exo组中肺转移的数量和大小显著增加(图4i和补充图4j、k),这验证了由外泌体let-7d-5p诱导的富含葡萄糖的肺部前转移灶作为一种有利的微环境,有利于循环肿瘤细胞(CTCs)的定植和转移灶的发展。
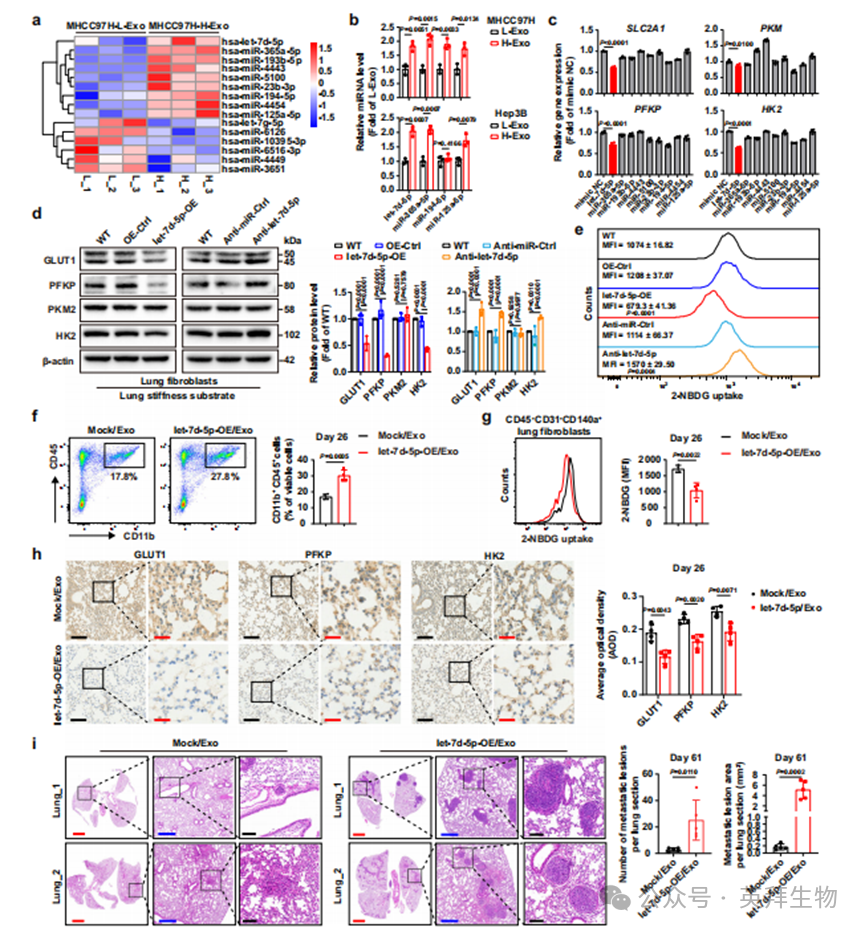
图4. 基质刚度调控的外泌体let-7d-5p促进肺转移前微环境形成过程中的葡萄糖富集
5.一个由基质刚度调节的外泌体let-7d-5p/HMGA2/E2F1乙酰化/GLUT1、PFKP和HK2通路参与调节肺成纤维细胞中的葡萄糖富集
为了阐明let-7d-5p调节肺成纤维细胞葡萄糖代谢的分子机制,我们使用了三种公开的生物信息学工具,包括TargetScan、miRWalk和miRTarBase,筛选了let-7d-5p的共同候选靶蛋白,并获得了154种共同候选蛋白(图5a)。在这些候选蛋白中,我们确定HMGA2是let-7d-5p的靶蛋白,原因如下:(1)在let-7d-5p靶蛋白的预测分析中,HMGA2的得分最高。(2)TCGA数据库分析显示,在肺和肝癌组织中,HMGA2与糖酵解途径呈正相关(补充图5a)。鉴于进入受体细胞(肺成纤维细胞)的外泌体let-7d-5p相当于受体细胞中let-7d-5p的上调,我们随后用慢病毒感染肺成纤维细胞,以确定let-7d-5p上调或下调对HMGA2表达的影响。结果表明,let-7d-5p的过表达显著抑制了HMGA2的蛋白表达,而let-7d-5p的敲低则增加了HMGA2的蛋白表达(图5b),这表明let-7d-5p与HMGA2之间存在负向调控关系。此外,根据TargetScan人类版本8.0的数据,我们观察到HMGA2 mRNA的3'UTR中有七个let-7d-5p的假定结合区域,所有结合区域都含有共同的序列(图5c)。随后,我们进行了荧光素酶报告基因实验,以明确let-7d-5p是否特异性地结合到HMGA2 mRNA的3'UTR。图5c中的结果显示,与对照组相比,共转染let-7d-5p和HMGA2 3'UTR-WT的荧光素酶活性明显降低,而HMGA2 3'UTR-mut组的荧光素酶活性保持不变(图5c),这表明let-7d-5p特异性地结合到HMGA2 mRNA的3'UTR,并调节其蛋白表达。这些数据证实了HMGA2是let-7d-5p的靶蛋白。我们进一步将HMGA2-OE质粒转染到let-7d-5p过表达的肺成纤维细胞中,以验证let-7d-5p是否通过HMGA2调节肺成纤维细胞的葡萄糖代谢。正如预期的那样,HMGA2的过表达显著增加了对照细胞中GLUT1、PFKP和HK2的表达,并挽救了let-7d-5p过表达所耗尽的这些蛋白的水平(图5d)。
HMGA2作为结构转录调节因子,调节包括E2F1在内的某些转录因子的活性,并且HMGA2与Rb相互作用可增强E2F1在垂体腺瘤中的乙酰化和活性。因此,我们推测外泌体let-7d-5p可能通过影响HDAC1从Rb-E2F1复合物的解离来调节肺成纤维细胞中E2F1的乙酰化。我们通过免疫沉淀实验确认了HMGA2和Rb在肺成纤维细胞核蛋白中的相互作用(图5e)。然后,我们提取了let-7d-5p过表达或下调的肺成纤维细胞的核蛋白(补充图5b),并以Rb为诱饵蛋白来捕获HMGA2、HDAC1和E2F1。图5f的结果显示,在let-7d-5p过表达的肺成纤维细胞中,HDAC1与Rb的相互作用更多,而在let-7d-5p下调的细胞中则较少。同时,HMGA2能够与HDAC1竞争结合Rb,但对E2F1与Rb的结合影响较小(图5f)。此外,我们进行了定量实验(ELISA),以特异性检测在用Rb抗体进行免疫沉淀后捕获的蛋白中HDAC1的蛋白水平。我们发现,在let-7d-5p-OE的肺成纤维细胞中,与Rb相互作用的HDAC1水平明显增加,而在抗let-7d-5p的肺成纤维细胞中,与Rb相互作用的HDAC1水平显著降低(补充图5c),这表明HMGA2的上调增加了HDAC1从Rb-E2F1复合物的解离。为了进一步验证let-7d-5p是否通过HMGA2调节E2F1的乙酰化,我们对let-7d-5p过表达或下调的肺成纤维细胞的核蛋白进行免疫沉淀实验,以捕获E2F1并检查E2F1的乙酰化水平。观察到在let-7d-5p过表达的肺成纤维细胞中,与E2F1相互作用的HDAC1更多,且E2F1的乙酰化水平较低(图5g)。相反,在let-7d-5p下调的肺成纤维细胞中,与E2F1相互作用的HDAC1较少,且E2F1的乙酰化水平增加(图5g)。我们进一步应用免疫荧光共定位分析来确认肺成纤维细胞核中HDAC1与E2F1之间的相互作用。结果显示,在let-7d-5p过表达的肺成纤维细胞中,与E2F1共定位的HDAC1更多,而在let-7d-5p下调的细胞中,与E2F1共定位的HDAC1较少(补充图5d)。这些数据表明,let-7d-5p的上调确实增加了HDAC1与E2F1的结合,从而降低了E2F1的乙酰化水平。转录因子通常通过结合其启动子序列来调节靶基因的表达。通过JASPAR数据库分析,我们预测E2F1是结合包括SLC2A1、PFKP和HK2在内的三个糖酵解相关基因启动子区域的潜在转录因子。通过进行ChIP-qPCR,我们确认了在肺成纤维细胞中E2F1在SLC2A1、PFKP和HK2的启动子上的结合增加(图5h和补充图5e)。
为了进一步阐明E2F1乙酰化在调节GLUT1、PFKP和HK2表达中的作用,我们用HDAC1抑制剂TSA处理let-7d-5p过表达的肺成纤维细胞。TSA干预明显挽救了let-7d-5p过表达引起的E2F1乙酰化水平的下降(图5i),并恢复了靶蛋白GLUT1、PFKP和HK2的表达(图5i)。接下来,使用在线乙酰化位点预测工具GPS-PAIL,鉴定出三个赖氨酸(K)残基位点,包括K117、K120和K125,是E2F1的主要乙酰化位点(补充图5f)。我们将Flag-E2F1(WT)-OE-质粒或Flag-E2F1(K117/120/125R)-OE-质粒分别转染到let-7d-5p下调或对照细胞的肺成纤维细胞中,并确认了细胞中E2F1蛋白的过表达(补充图5g)。随后的免疫沉淀分析显示,无论是在let-7d-5p下调组还是对照组中,带有Flag-E2F1(K117/120/125R)OE-质粒的肺成纤维细胞中E2F1的乙酰化水平显著低于带有Flag-E2F1(WT)-OE-质粒的肺成纤维细胞(图5j)。更重要的是,K117/120/125R突变明显逆转了let-7d-5p敲低引起的靶蛋白(GLUT1、PFKP和HK2)的上调(图5k)。因此,K117、K120和K125确实是E2F1的主要乙酰化位点,其乙酰化水平影响靶蛋白的表达。综上所述,所有上述证据表明,一个由基质刚度调节的外泌体let-7d-5p/HMGA2/E2F1乙酰化/GLUT1、PFKP和HK2通路参与调节肺成纤维细胞中的葡萄糖富集。
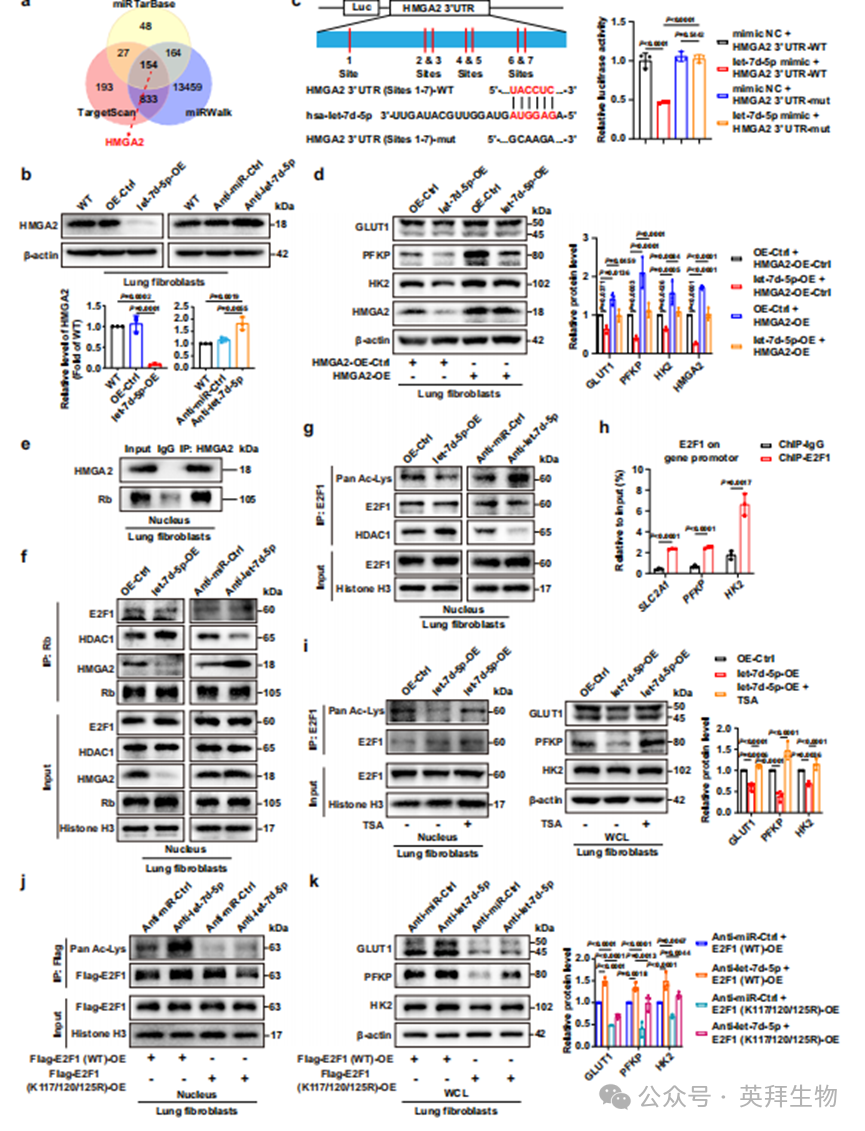
图5. 一个由基质刚度调节的外泌体let-7d-5p/HMGA2/E2F1乙酰化/GLUT1、PFKP和HK2通路参与调节肺成纤维细胞中的葡萄糖富集
6.基质刚度调节的外泌体miR-365a-5p有效调节血管生成和血管通透性以影响肺部前转移灶中的葡萄糖富集
除了肺成纤维细胞减少葡萄糖消耗对葡萄糖富集的贡献外,血管生成和血管通透性作为前转移灶的关键特征,也通过增加葡萄糖供应来影响葡萄糖富集。基于上述鉴定的差异miRNA,我们继续确定基质刚度调节的外泌体中哪种miRNA在肺部前转移灶形成过程中有效控制血管生成和血管通透性。我们将9种上调的miRNA模拟物分别转染到人脐静脉内皮细胞(HUVECs)中,并分析了不同miRNA对血管生成和血管通透性的贡献。只有miR-365a-5p明显抑制了HUVECs中TJP1(ZO-1的基因名称)和CDH5(VE-钙粘蛋白的基因名称)的表达(图6a)。此外,miR-365a-5p模拟物显著降低了ZO-1和VE-钙粘蛋白的蛋白水平,但提高了VEGFR2的蛋白水平,而miR-365a-5p抑制剂则表现出相反的效果(图6b和补充图6a)。我们进一步分析了miR-365a-5p是否破坏内皮屏障完整性并促进癌细胞外渗(图6c),发现异位表达miR-365a-5p的HUVEC单层对FITC-葡聚糖和MHCC97H-GFP细胞的通透性更高(图6d、e)。相反,HUVECs中miR-365a-5p的敲低导致内皮渗漏明显减少(图6d、e)。此外,我们进行了体外绒毛尿囊膜(CAM)实验和体外管状形成实验,以阐明miR-365a-5p在血管生成中的作用。miR-365-1-5p(小鼠miR-365-1-5p的序列与人类miR-365a-5p相同)在Hepa1-6细胞中稳定过表达(补充图6b),并收集其培养上清进行CAM实验。与用Hepa1-6-Mock/CM处理的对照组相比,用Hepa1-6-miR-365-1-5p-OE/CM处理的CAM显示出血管数量显著增加(图6f)。在体外管状形成实验中,HUVECs中miR-365a-5p的异位表达也增加了管状数量,而miR-365a-5p的敲低则减少了管状数量(图6g)。这些结果验证了来自肝细胞癌(HCC)细胞的miR-365a-5p(miR-365-1-5p)足以教育血管内皮细胞并诱导血管通透性和血管生成。随后,我们进行了体内的FITC-葡聚糖通透性实验,以评估外泌体miR-365a-5p对血管通透性的影响。我们从Hepa1-6-Mock和Hepa1-6-miR-365-1-5p-OE细胞的培养上清中分离并纯化了外泌体,随后确认了纯化外泌体中miR-365-1-5p的上调(补充图6b)以及纯化外泌体的质量(补充图6c、d)。外泌体通过尾静脉每两天注射一次,持续26天(补充图6e)。与PBS或Mock/Exo组相比,miR-365-1-5p-OE/Exo组中的FITC-葡聚糖更容易从肺血管渗出(图6h、i),这意味着外泌体miR-365-1-5p可以通过教育血管内皮细胞来破坏肺血管的完整性。同时,miR-365-1-5p-OE/Exo组的肺组织中CD31的表达增强(图6j、k)。上述结果表明,外泌体miR-365a-5p(miR-365-1-5p)有助于肺部前转移灶中的血管生成和血管通透性。
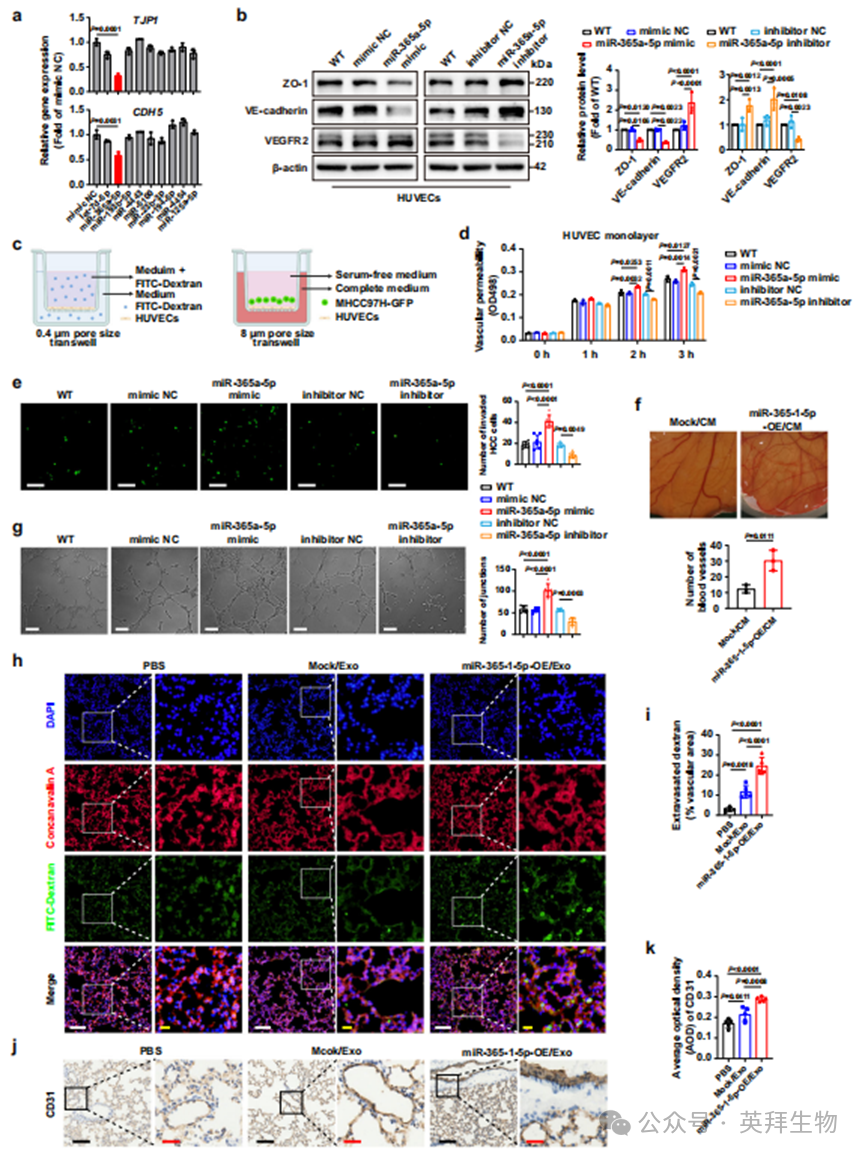
图6. 基质刚度调节的外泌体miR-365a-5p有效调节血管生成和血管通透性以影响肺部前转移灶中的葡萄糖富集
7.外泌体miR-365a-5p通过抑制TRPC4AP/Ca2+/CaMKII/ERK5/KLF2/4通路增加血管生成和血管通透性
利用三种公开的生物信息学工具(TargetScan、miRWalk和miRTarBase),我们获得了97种miR-365a-5p的共同靶蛋白(图7a)。在这些候选靶蛋白中,由于钙离子内流对血管功能的巨大影响,以及TRPC4AP与TJP1(ZO-1的基因名称)或CDH5(VE-钙粘蛋白的基因名称)在TCGA分析中的正相关性,TRPC4AP被选为miR-365a-5p的靶蛋白(补充图7a)。如图7b所示,miR-365a-5p模拟物显著下调了HUVECs中TRPC4AP的表达,而miR-365a-5p抑制剂则显著上调了HUVECs中TRPC4AP的表达,表明miR-365a-5p与其靶蛋白TRPC4AP之间存在负向调控关系。此外,荧光素酶报告基因实验显示,共转染TRPC4AP 3'UTR-WT和miR-365a-5p模拟物的细胞中荧光素酶活性显著降低,而共转染TRPC4AP 3'UTR-mut和miR-365a-5p模拟物的细胞中荧光素酶活性保持不变(图7c),进一步验证了miR-365a-5p在TRPC4AP表达中的特定调控作用。TRPC4AP是一种与钙离子通道相关的蛋白,可与TRPC4相互作用以增强钙库操作性Ca2+内流并增加细胞内钙离子浓度。为了解开miR-365a-5p是否调节TRPC4AP诱导的Ca2+内流,我们使用对Ca2+敏感的荧光染料Fluo-4 AM检测了转染miR-365a-5p模拟物或抑制剂的HUVECs中的Ca2+内流。我们发现miR-365a-5p模拟物显著降低了HUVECs中细胞内Ca2+的荧光强度,而miR-365a-5p抑制剂则明显提高了荧光强度(图7d和补充图7b),这表明miR-365a-5p能够抑制TRPC4AP的表达以减少HUVECs中的钙离子内流。随着细胞内钙离子浓度的增加,CaMKII(一种多功能的丝氨酸/苏氨酸激酶)通常被激活以磷酸化下游分子,如MAPK、AMPK、AKT和Src。在一篇报道中,抑制CaMKII可以逆转Piezo1通过MEKK3/MEK5/ERK5信号通路在原代HUVECs中诱导的KLF2/4表达。KLF2通常通过抑制VEGFR2的表达来负向调节血管生成,而KLF4则通过激活ZO-1和VE-钙粘蛋白的表达来维持血管完整性。因此,我们推测miR-365a-5p通过抑制CaMKII/ERK5通路逆转了TRPC4AP激活的KLF2/4表达。我们检测了转染miR-365a-5p模拟物或抑制剂的HUVECs中CaMKII/ERK5/KLF2/4的激活状态,发现miR-365a-5p模拟物显著抑制了CaMKII和ERK5的磷酸化水平以及KLF2和KLF4的表达水平(图7e)。相反,miR-365a-5p抑制剂明显逆转了上述变化(图7e),这表明miR-365a-5p确实通过抑制TRPC4AP来破坏CaMKII/ERK5/KLF2/4的激活。接下来,我们将TRPC4AP过表达质粒转染到miR-365a-5p模拟物转染的HUVECs中,并进一步确定miR-365a-5p是否通过抑制TRPC4AP/Ca2+/CaMKII/ERK5/KLF2/4通路来增加血管生成和血管通透性。如图7f所示,miR-365a-5p模拟物显著增加了VEGFR2的表达,降低了ZO-1和VE-钙粘蛋白的表达,而TRPC4AP的上调阻止了miR-365a-5p在CaMKII/ERK5/KLF2/4通路中的失活作用(图7f)。值得注意的是,KLF4的沉默减弱了miR-365a-5p抑制剂对ZO-1和VE-钙粘蛋白表达的促进作用(图7g和补充图7c),而KLF2的沉默则逆转了miR-365a-5p抑制剂对VEGFR2表达的抑制作用(图7h和补充图7c)。综上所述,外泌体miR-365a-5p通过抑制TRPC4AP的表达来失活CaMKII/ERK5/KLF2/4信号通路,从而促进血管生成和血管通透性。
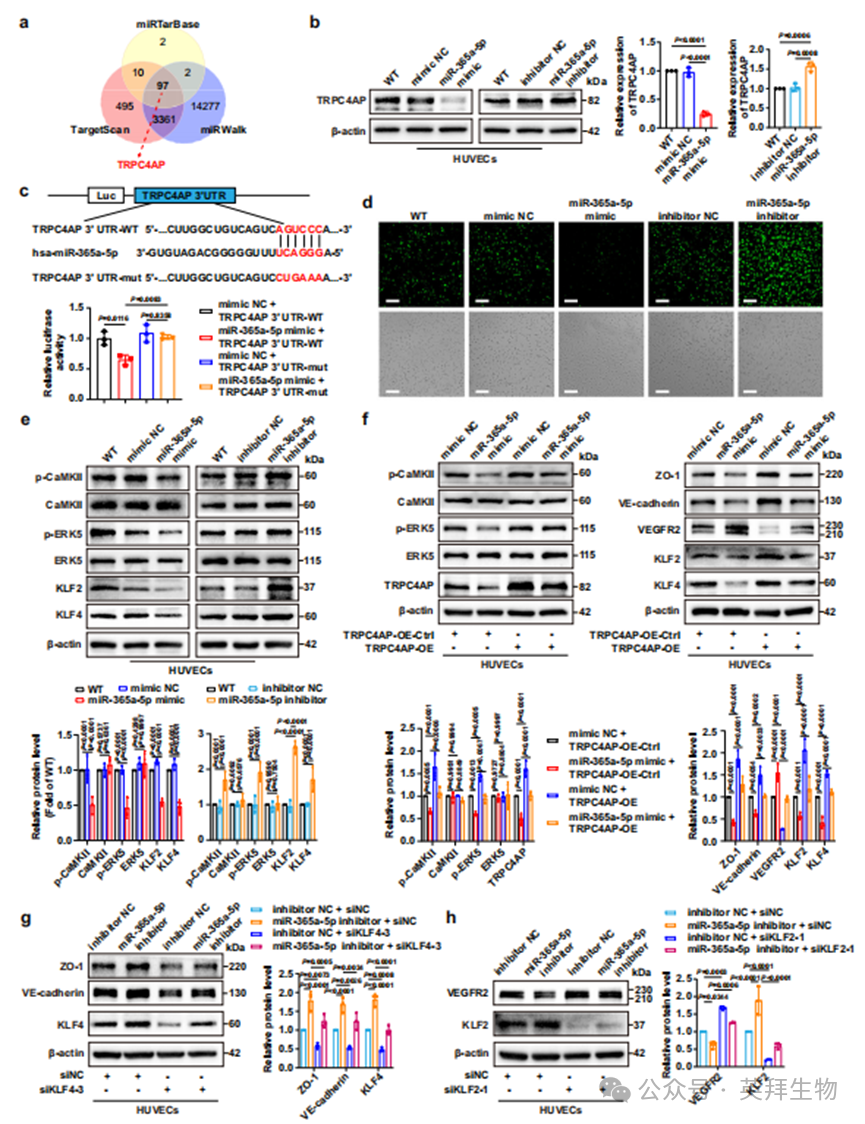
图7. 外泌体miR-365a-5p通过抑制TRPC4AP/Ca2+/CaMKII/ERK5/KLF2/4通路增加血管生成和血管通透性
8.COL1高/LOX高与let-7d-5p和miR-365a-5p在肝细胞癌组织中的关联及其临床意义
基质变硬主要是由于细胞外基质蛋白(如I型胶原蛋白COL1)的过度沉积和交联所致。赖氨酰氧化酶(LOX)可以有效增强细胞外基质蛋白的交联水平。因此,COL1和LOX的表达水平常被用来表示肝脏基质硬度的等级。以肿瘤组织中COL1和LOX的中位表达水平为阈值,将肝细胞癌(HCC)患者分为两组:COL1低/LOX低组(低硬度组,24例)和COL1高/LOX高组(高硬度组,24例)。我们采用荧光原位杂交(FISH)实验检测肝细胞癌组织中let-7d-5p和miR-365a-5p的表达,发现COL1高/LOX高组中let-7d-5p和miR-365a-5p的表达水平均明显高于COL1低/LOX低组(图8a、b),表明基质硬度与let-7d-5p或miR-365a-5p表达呈正相关(图8a、b)。接下来,我们分析了COL1/LOX表达与肿瘤大小、微血管侵犯、肿瘤分化和肿瘤复发等临床病理指标的关联(表1),发现COL1高/LOX高组的肝细胞癌组织表现出更高的let-7d-5p和miR-365a-5p水平以及更差的肿瘤分化,COL1高/LOX高组的肝细胞癌患者肿瘤复发率更高。生存分析显示,COL1高/LOX高组的肝细胞癌患者总生存期(OS)更差,无病生存期(DFS)也较差(图8c)。基于let-7d-5p和miR-365a-5p的中位表达水平,我们进一步将肝细胞癌患者分为两组,包括let-7d-5p低/miR-365a-5p低组(16例)和let-7d-5p高/miR-365a-5p高组(16例)。我们发现let-7d-5p高/miR-365a-5p高组的肝细胞癌患者在生存分析中表现出不利的OS和DFS(图8d)。在TCGA肝癌患者队列中,我们还发现let-7d高/miR-365a高提示不利的OS和DFS(补充图8a、b)。因此,这些数据表明,高基质硬度与肝细胞癌组织中let-7d-5p或miR-365a-5p表达增加密切相关,与细胞实验中的发现一致。高基质硬度和let-7d-5p/miR-365a-5p高表达可以更好地预测肝细胞癌患者的不良预后和肿瘤复发。
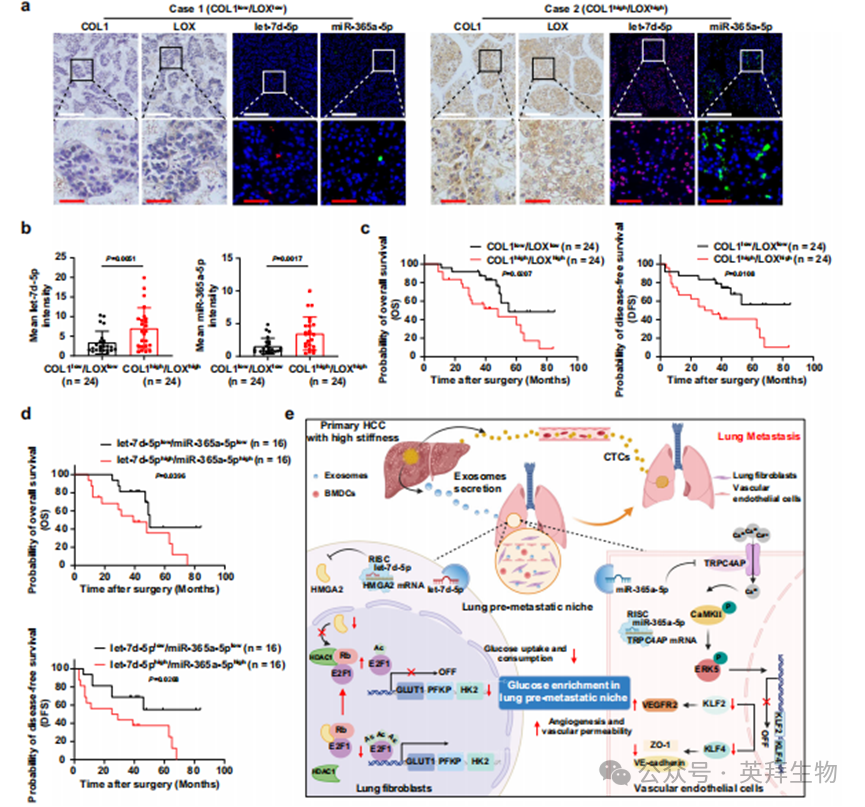
图8. COL1高/LOX高与let-7d-5p和miR-365a-5p在肝细胞癌组织中的关联及其临床意义
结论:
该研究揭示了肝细胞癌(HCC)中紧密连接蛋白4(CLDN4)的棕榈酰化通过促进肝细胞向胆管细胞谱系转化(HBT)和激活Notch信号通路,导致仑伐替尼耐药的分子机制。棕榈酰化位点C104和C107由zDHHC5催化,增强了CLDN4在脂筏中的锚定,抑制了其内吞作用,从而维持其在细胞膜上的稳定表达。棕榈酰化的CLDN4将接触蛋白1(CNTN1)募集到脂筏中,激活Notch信号通路,上调胆管细胞谱系标志物,下调肝细胞谱系标志物,促进HBT。CLDN4的高表达与仑伐替尼耐药性密切相关。Salvianolic acid B(SalB)通过抑制zDHHC5,降低CLDN4的棕榈酰化水平,减少HBT,恢复仑伐替尼的敏感性。联合化疗(如吉西他滨/顺铂)对发生HBT的HCC患者可能有效。
参考文献:
Zhao Y, Yu H, Li J, Qian J, Li M, Zhang X, Wang M, Wang Y, Dong Y, You Y, Zhou Q, Gao D, Zhao Y, Liu B, Chen R, Ren Z, Wang Z, Zhang K, Cui J. A glucose-enriched lung pre-metastatic niche triggered by matrix stiffness-tuned exosomal miRNAs in hepatocellular carcinoma. Nat Commun. 2025 Feb 18;16(1):1736.

本周
本月
本年
LetPub发布最新SCI影响因子查询及期刊投稿分析系统
ELISpot试剂盒限时特惠!一口价低至500元,加赠免费读板服务
草甘膦(glyphosate)酶联免疫分析(ELISA)试剂盒使用说明书
应用gentleMACS™灌流技术从脂肪肝小鼠模型高效分离肝细胞与非实质细胞
单个细胞也能提取核酸?超全干货教你微量样本发高分(含完整电子版宝典资料))
链脲佐菌素 (Streptozotocin,STZ)-糖尿病动物模型造模
LetPub完整SCI影响因子、期刊分区查询系统
研究思路 | 多组学专题——如何进行转录组+蛋白组关联分析?
Elabscience® 从原料到标记,打造属于中国自己的流式抗体品牌
淋巴细胞百分比偏低原因解析
- 促销公告
- 更多 ›
你可能感兴趣的产品
- miProfile人类肝细胞癌外泌体miRNA qPCR阵列/miProfile人类肝细胞癌外泌体miRNA qPCR阵列/miProfile人类肝细胞癌外泌体miRNA qPCR阵列
- 间充质基质细胞外泌体包封miRNA-101
- FyMeso人间充质干细胞无血清培养基(chemical defined)
- 冷冻研磨仪/低温冷冻研磨仪/组织研磨仪
- 美国PE耗材配件珀金埃尔默GC色谱耗材
- 外泌体提取纯化试剂盒(组织、细胞上清、血液、尿液等)
- RNA快速提取试剂盒(带DNA清除柱 )
- BVC professional 真空吸液系统(废液处理器)
- 链酶蛋白酶浓缩液(20X)
- 氘灯Waters 486 D2 lamp 2000时
- Merck默克化学试剂一级代理
- 感光细胞转录因子TULP3抗体
