


1. 加入 1/10 体积的乙酸钠(3 mol/L ,PH=5.2)于 DNA 溶液中充分混匀,使其 最终浓度为 0.3 mol/L;2. 加入 2 倍体积用冰预冷的乙醇混合后再次充分混匀置于- 20℃中 15~30 分钟;3. 12,000 g 离心 10 分钟,小心移出上清液,吸去管壁上所有的液滴;4. 加入 1/2 离心管容量的 70%乙醇,12000g 离心 2 分钟,小心移出上清液,吸去管壁上所有的液滴;5. 于室温下将开盖的 EP 管的置于实验桌上以使残留的液体挥发至干;6. 加适量的 ddH2O 溶解 DNA 沉淀。

TRIzol试剂适用于从细胞和组织中快速分离RNA。TRIzol试剂有多组分分离作用,与其他方法如硫氰酸胍/酚法、酚/SDS法、盐酸胍法、硫氰酸胍法等相比,最大特点是可同时分离一个样品的RNA\DNA\蛋白质.TRIzol使样品匀浆化,细胞裂解,溶解细胞内含物,同时因含有RNase抑制剂可保持RNA的完整性。在加入氯仿离心后,溶液分为水相和有机相,RNA在水相中。取出水相用异丙醇沉淀可回收RNA;用乙醇沉淀中间层可回收DNA;用异丙醇沉淀有机相可回收蛋白质。TRIzol试剂可用于小量样品(50~100mg组织、5×106细胞)也适用于大量样品(≥1g组织、>107细胞)。对人,动物,植物组织,细菌均适用,可同时处理大量不同样品,整个提取过程在一小时内即可完成。分离的总RNA无蛋白质和DNA污染,可用于Northern Blot,dot blot,ployA筛选,体外翻译,RNase保护分析和分子克隆。在用于RT-PCR时如果两条引物存在于一个单一外显子内,建议用无RNase的DNaseⅠ处理RNA样品,避免出现假阳性。共纯化的DNA可用作标准,比较不同样品RNA的得率,也可用于P

一、IP前的准备工作:coIP:分为内源性蛋白的相互作用和非内源性蛋白相互作用(后者包括外源蛋白之间以及外源拖内源蛋白)。非内源性蛋白的相互作用,主要是通过过表达来实现,通常为简化实验、提高IP效率会让外源蛋白带上标签(tagged;这也是没有相应的可用于IP的内源蛋白抗体之前唯一可行的方案),因此就tag的使用而言存在很多小技巧。1. His-tagged主要用于纯化蛋白。有些人喜欢用His-tagged蛋白然后进行coIP,实际上它存在潜在的隐患。当初鄙人用Sigma a-His(鼠单抗;免费广告)WB外源蛋白发现CE里面一塌糊涂(带型漂亮就是非常多的带),那时候还抱怨这个抗体特异性不好。后来,当了解到很多蛋白会有富含histidine的domain以后,恍然大悟,恰恰是因为效价非常好,所以很多内源性的蛋白都被该抗体识别。同理,如果用Ni-NTA beads去PD His-tagged的蛋白,完全有可能PD那些内源性的富含histidine domain的蛋白,造成coIP的假象,而这样的蛋白还非常多。2. tandem-tagged不能用于分析钙调信号通路。tandem tag实
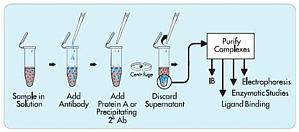
IP是利用抗原蛋白质和抗体的特异性结合以及细菌蛋白质的“protein A/G"特异性地结合到抗体(免疫球蛋白)的FC片段的现象开发出来的方法。目前多用protein A/G预先结合在argarose beads上,使之与含有抗原的溶液及抗体反应后,beads上的prorein A/G就能达到吸附抗原的目的。通过低速离心,可以从含有目的抗原的溶液中将目的抗原与其它抗原分离。 免疫沉淀实验的操作步骤比较多,同时由于在非变性条件下进行实验,所以要得到一个完美的实验结果,不仅需要高质量的抗体,同时对免疫沉淀体系也需要有严格的控制指标。免疫沉淀实验从:蛋白样品处理;抗体-agarose beads孵育;抗体-agarose beads复合物洗涤到最后的鉴定,每步都非常关键,需要严格控制实验流程中每个关键步骤的质量,才能最终达到你的实验目的。 IP实验步骤 基本实验步骤 1. 收获细胞,加入适量细胞IP裂解缓冲液(含蛋白酶抑制剂),冰上或者4℃裂解30 min, 12 000g离心30 min后取上清; 2. 取少量裂解液以备Western blot分析,剩余裂解液将1 μg 相应的抗体和

一、原理:免疫共沉淀(Co-Immunoprecipitation)是以抗体和抗原之间的专一性作用为基础的用于研究蛋白质相互作用的经典方法。是确定两种蛋白质在完整细胞内生理性相互作用的有效方法。其原理是:当细胞在非变性条件下被裂解时,完整细胞内存在的许多蛋白质-蛋白质间的相互作用被保留了下来。如果用蛋白质X的抗体免疫沉淀X,那么与X在体内结合的蛋白质Y也能沉淀下来。目前多用精制的prorein A预先结合固化在argarose的beads上,使之与含有抗原的溶液及抗体反应后,beads上的prorein A就能吸附抗原达到精制的目的。这种方法常用于测定两种目标蛋白质是否在体内结合;也可用于确定一种特定蛋白质的新的作用搭档。其优点为:(1)相互作用的蛋白质都是经翻译后修饰的,处于天然状态;(2)蛋白的相互作用是在自然状态下进行的,可以避免人为的影响;(3)可以分离得到天然状态的相互作用蛋白复合物。缺点为:(1)可能检测不到低亲和力和瞬间的蛋白质-蛋白质相互作用;(2)两种蛋白质的结合可能不是直接结合,而可能有第三者在中间起桥梁作用;(3)必须在实验前预测目的蛋白是什么,以选择最后检测的抗

这段时间我的 WB 取得了一些进展,终于有点时间好好地整理一下实验过程中得到的一些经验。我写这篇文章,是因为这些经验在此前我看过的所有文章中,都没有人提及过,但我确实觉得非常重要。我相信,对于很多跟我一样的 WB 新手们,会有一些帮助。好,废话不多说,先上两张图,让大家有一点直观的感受。相信看完这张图,大家已经大致猜到答案了。没错,第一张图是带有 Marker 的,而第二张图是剪掉 Marker 以后的。我们可以非常清晰地看到,前后两张图差别极其明显。第一张图只能隐隐约约地看到 3 个目的条带,而第二张图 6 个目的条带全部可以看到。而造成这样大反差的根本原因就在于 Marker 了。为什么会这样呢?有三个很重要的原因。第一,选择的抗体不是单克隆抗体。第二,该抗体和 Marker 的结合力太强了,远远超过了跟目的蛋白的结合力。第三,我们使用的成像系统并不能像人一样识别哪些是目的条带,哪些是非目的条带,为了保证成像区域不至于过度曝光,成像系统会严格地控制曝光时间,在特定的曝光时间下,尽管成像区域没有因为过度曝光而导致影像失常,但信号相对较弱的目的条带却因为没有足够的曝光时间而导致无法正常

PCR 是分子生物学科研人员必做的实验,为了扩增目的基因,经常要对 PCR 的体系进行“摸条件”,特别是一些 GC 含量高的目的基因或者 DNA 纯度不高的样本! “摸条件”成为 PCR 实验最费时费力的工作! 虽然市场上有很多 PCR 的 MasterMix 试剂,将酶、dNTP、反应缓冲液混合在一起,很方便客户的使用,但仅仅是简单的混合,极少公司对MIX的应用范围进行广泛的实验和优化,只能做一些不复杂的 PCR。 百泰克通过长时间、无数次的实验优化,终于推出了适用范围广、稳定性良好的PCRMIX,是目前最“傻瓜”的 PCR 反应体系之一,不论是质粒、菌液、CDNA、基因组 DNA、或者是直接裂解的粗提物,GC 含量高达 75% 的模板,都能轻松扩增,不需要费力的“摸条件”! 本产品包含酶、dNTP、特殊稳定剂和优化的反应缓冲液,浓度为 2x。DNA 扩增时,只需加入模板,引物和水,使 MasterMix 溶液的浓度为 1× 即可进行反应。具有快速、灵敏度高、特异性强、稳定性好等优点。进行 PCR 扩增可以减少操作时间,避免因多步操作带来的污染,特别适宜高通量筛选基因的扩增 ,Taq
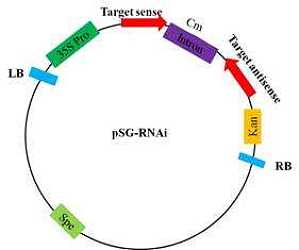
目前RNAi技术已经进入了生物科学研究的许多领域,成为了一种主要的生物学研究工具,发展得相当迅速,并受到了此次诺贝尔奖评审委员会 < language=Java1.1 src="/newsf/js/inpic.js">的青睐,获得2006年诺贝尔奖医学/生理奖。然而这一才历经十个年头的技术依然对于许多研究人员来说还是很陌生的,以下是有关RNAi技术在哺乳动物细胞中应用的具体设计策略,主要来自于《遗传》杂志2005年“RNA干涉(RNAi)技术应用于哺乳动物细胞的研究策略”,希望能给从事或者准备从事RNAi相关实验的研究人员以帮助。一、靶siRNA序列选择靶siRNAA序列选择是RNAi实验成败的关键。哺乳动物细胞RNAi实验中,使用最广泛且最有效的是21bp siRNA。siRNA由正义链和反义链组成,两条链3’端均有2个碱基突出,一般为UU或dTdT,其中正义链的前19nt与靶基因序列相同。1. siRNA设计的原则SiRNA双链设计时,一般在靶mRNA起始密码下游100—200bp至翻译终止密码上游d0—100bp的范围内搜寻AA序列,并记录每个AA3’端相邻19个核苷
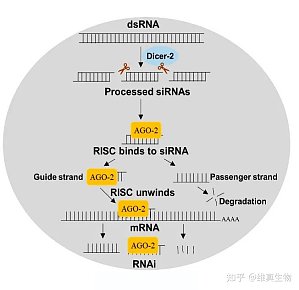
一、RNAi的分子机制通过生化和遗传学研究表明,RNA干扰包括起始阶段和效应阶段(inititation and effector steps)。在起始阶段,加入的小分子RNA被切割为21-23核苷酸长的小分子干扰RNA片段(small interfering RNAs, siRNAs)。证据表明;一个称为Dicer的酶,是RNase III家族中特异识别双链RNA的一员,它能以一种ATP依赖的方式逐步切割由外源导入或者由转基因,病毒感染等各种方式引入的双链RNA,切割将RNA降解为19-21bp的双链RNAs(siRNAs),每个片段的3’端都有2个碱基突出。在RNAi效应阶段,siRNA双链结合一个核酶复合物从而形成所谓RNA诱导沉默复合物(RNA-induced silencing complex, RISC)。激活RISC需要一个ATP依赖的将小分子RNA解双链的过程。激活的RISC通过碱基配对定位到同源mRNA转录本上,并在距离siRNA3’端12个碱基的位置切割mRNA。尽管切割的确切机制尚不明了,但每个RISC都包含一个siRNA和一个不同于Dicer的RNA酶。另外,还
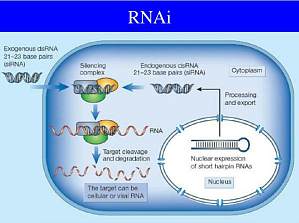
近年来的研究表明,一些短片断的双链RNA可以通过促使特定基因的mRNA降解来高效、特异的阻断体内特定基因表达,诱使细胞表现出特定基因缺失的表型, 称为RNA干扰(RNA interference,RNAi).siRNA(small interfering RNAs)就是这种短片断双链RNA分子,能够以序列同源互补的mRNA为靶目标,降解特定的mRNA.RNAi的发现具有划时代的意义,它不仅深入揭示 了细胞内基因沉默的机制,而且它还是后基因组时代基因功能分析的有力工具,极大地促进了人类揭示生命奥秘的进程.现在越来越多的研究人员开始采用RNAi 来研究生物体的基因表达.RNAi技术可广泛应用到包括功能基因组学,药物靶点筛选,细胞信号传导通路分析,疾病治疗等等.目前为止较为常用的5种制备siRNAs的方法包括:·化学合成·体外转录·长片断dsRNAs经RNase III 类降解 (e.g. Dicer, E. coli, RNase III)·siRNA表达载体或者病毒载体在细胞中表达siRNAs·PCR制备的siRNA表达框在细胞中表达获得高纯度的siRNA产物是进行实验的第一步,而转染的

1. 收到细胞株包裹时,请检查细胞株冷冻管是否有解冻情形,若有请立即通知。细胞株请尽速开始培养,或立即冷冻保存(置于-70 ℃,隔夜后,移到液氮)。2. 冷冻细胞解冻程序:2.1. 依据细胞株数据单指定之基础培养基种类、血清种类和其它指定之成份和比例, 制备培养基。绝大多数之细胞均无法立即适应不同之基础培养基或不同之 血清种类,若因实验需要,必须有所不同时,务必以缓慢比例渐次改变培养基组成, 确定细胞适应后, 方进行所需之实验。2.2. FBS (fetal bovine serum,胚牛血清),CS (calf serum,小牛血清)和HS (horse serum,马血清),对细胞而言差异极大,请务必依据细胞株资料单指定之血清种类培养之。2.3. 将培养基置于37 ℃ 水槽中回温,回温后喷以70 % 酒精并擦拭之,移入无菌操作台内。取出冷冻管,立即放入37 ℃ 水槽中快速解冻,水面高度不可接近或高过冷冻管之盖沿,否则易发生污染。轻摇冷冻管使其在1 分钟内全部融化后,以70 % ethanol 擦拭冷冻管外部, 移入无菌操作台内。2.4. 依据细胞种类和浓度,于无菌操作台内取10 m
原代细胞冻存技术:1、选用生长情况好,数量较多的原代细胞,冻存前一天换液一次。2、贴壁细胞需用0.25%胰酶常规消化将细胞消化下来,将细胞悬液收集至离心管中。3、1000rpm离心5分钟,弃上清液。4、沉淀加含DMSO的培养液,计数,调整至(1-10)×106 /ml。5、将悬液分至冻存管中,每管1 ml。6、密封冻存管,封口一定要严,否则复苏时易出现爆裂。7、用记号笔标明细胞种类,冻存日期。8、按下列顺序降温:室温→4℃(20分钟〕→冰箱冷冻室(30分钟)→超低温冰箱(-80℃过夜)→液氮。
做实验前要先学会冻存,以备自己在后面的实验中能保持同一来源、稳定可靠的细胞,且可减少污染或其它不利影响。1、影响冻存细胞活性的因素(1)细胞冻存保护剂:常用的有二甲基亚砜(DMSO)和甘油,常用的浓度为5%-10%(90%小牛血清)。DMSO对二倍体细胞的毒性较甘油小。(2)细胞悬液的冻结速度:慢冻时冰冻在细胞外形成,因此不致损害细胞。多数细胞以每分钟下降1℃的速度,降至-20℃冻结。均可获得满意效果。(3)保存温度:液氮罐,可达到-196℃,此时所有的物理化学活动均降至最低限度,以保证细胞长期保存。(4)复苏:急速融化复苏可防止损害细胞。冻结细胞从液氮中取出后,应立即放入37℃~43℃的水浴中,30秒至一分钟内融化。2、冻存与复苏方法适用于原始组织、原代细胞和细胞系(株)。(1)细胞用胰蛋白酶+EDTA消化后用冻存液稀至2~5X106/ml。(2)分装于2ml冻存管中,装量1ml,封口,作好标记,用几层纱布扎好。(3) 冻存管进入液氮前应有一个渐降温的过程,以1℃/min的速度降温,一般挂在液氮上空8小时后,投入液氮贮存罐中长期保存。(4)为使冻存的细胞复苏,将冻存管置于37℃~43
1、欲冷冻保存之细胞应在生长良好(log phase) 且存活率高之状态,约为80 – 90 %致密度。2、冷冻前检测细胞是否仍保有其特有性质。例如hybridoma 应在冷冻保存前一至二日测试是否有抗体之产生。3、注意冷冻保护剂之品质。DMSO 应为试剂级等级,无菌且无色(以0.22 micron FGLP Telflon 过滤或是直接购买无菌产品,如Sigma D-2650),以5~10 ml 小体积分装,4 oC 避光保存,勿作多次解冻。Glycerol 亦应为试剂级等级,以高压蒸汽灭菌后避光保存。在开启后一年内使用,因长期储存后对细胞会有毒性。4、冷冻保存之细胞浓度:(1)normal human fibroblast: 1~3 x 106 cells/ml(2)hybridoma: 1~3 x 106 cells/ml,细胞浓度不要太高,某些hybridoma 会因冷冻浓度太高而在解冻24 小时后死去。(3)adherent tumor lines: 5~7 x 106,依细胞种类而异。Adenocarcinoma 解冻后须较高之浓度,而HeLa 只需1-3 x 106 ce

浅谈流式核内蛋白染色步骤:1、染表面抗体 按说明书条件孵育全血样品孵育抗体后加入不含固定剂的溶血素(按厂家说明操作)溶血完毕。2、用2ml预冷流式染色缓冲液(Flow Cytometry Staining Buffer)洗涤细胞300-400g离心5分钟,去上清。3、用3倍体积的固定/破膜稀释液(#00-5223)稀释固定/破膜浓缩液(#00-5123)制成固定/破膜工作液,注意要新鲜配制,每个样本的用量为1ml 。4、旋涡震荡重悬细胞后加入1ml的固定/破膜工作液(1:3稀释好)再次旋涡混匀,避光4℃孵育30分钟到60分钟。5、将破膜缓冲液(#00-8333)用去离子水1:9稀释,制成工作液(每个样本的用量为4~5ml)。6、无需洗涤,直接加入2ml破膜缓冲液工作液(1:9稀释好)300-400g离心洗涤细胞并弃去上清液(可选)重复洗涤一次。7、加入100 ul流式染色缓冲液重悬细胞体系中加入2 ul 胞内抗体来源的血清,室温避光孵育30分钟。8、体系加入适量的核内(及胞浆)抗原抗体,对照管加入等量的同型对照,按说明书条件孵育。9、加入2ml破膜缓冲液工作液离心洗涤细胞并弃去上清液(
一、原理1、E .coli 表达系统E .coli 是重要的原核表达体系。在重组基因转化入E .coli 菌株以后,通过温度的控制,诱导其在宿主菌内表达目的蛋白质,将表达样品进行SDS-PAGE 以检测表达蛋白质。2、外源基因的诱导表达提高外源基因表达水平的基本手段之一,就是将宿主菌的生长与外源基因的表达分成两个阶段,以减轻宿主菌的负荷。常用的有温度诱导和药物诱导。本实验采用异丙基硫代-β-D-半乳糖昔(IPTG)诱导外源基因表达。不同的表达质粒表达方法并不完全相同,因启动子不同,诱导表达要根据具体情况而定。二、材料1、诱导表达材料(1 )LB (Luria—Bertani))培养基酵母膏 (Yeast extract)5g 蛋白胨 (Peptone)10gNaCl10g 琼脂 (Agar)1-2%蒸馏水 (Distilled water)1000ml pH 7.0适用范围:大肠杆菌(2 )IPTG 贮备液:2 g IPTG溶于10 ml 蒸馏水中,0 .22 μm 滤膜过滤除菌,分装成1 ml /份,-20 ℃ 保存。(3 )l× 凝胶电泳加样缓冲液:50 mmol / L Tris
一、SP 法1)脱蜡、水化;2)PBS 洗 2~3 次各 5 分钟;3)3% H2O2(80% 甲醇)滴加在 TMA 上,室温静置 10 分钟;4)PBS 洗 2~3 次各 5 分钟;5)抗原修复;6)PBS 洗 2~3 次各5分钟;7)滴加正常山羊血清封闭液,室温 20 分钟。甩去多余液体。8)滴加Ⅰ抗 50μl,室温静置 1 小时或者4℃过夜或者37℃1小时。9)4℃ 过夜后需在37℃复温45分钟。10)PBS 洗 3 次各 5 分钟;11)滴加 Ⅱ 抗 40~50 μl,室温静置,或 37℃ 1 小时;12)II抗中可加入 0.05% 的 tween-20。13)PBS 洗 3 次各 5 分钟;14)DAB 显色 5~10 分钟,在显微镜下掌握染色程度;15)PBS或自来水冲洗 10 分钟;16)苏木精复染 2 分钟,盐酸酒精分化;17)自来水冲洗 10~15 分钟;18)脱水、透明、封片、镜检。二、SABC法1)脱蜡、水化。2)PBS 洗两次各 5 分钟。3)用蒸馏水或PBS配置新鲜的3%H2O2,室温封闭5~10分钟,蒸馏水洗3次。4)抗原修复。5)PBS洗5分钟。6)滴加正
ELISA检测系统可谓是免疫学反应应用到科研生产中最为广泛最为灵敏的技术手段。可是在新老手操作过程中总是会出现或大或小的问题,本人在刚开始做ELISA时就面临很多困难,虽然有师兄师姐铺路,但还是常常做得一塌糊涂,比如说花板,假阳性,全显色,全部显色,显色比空白还低......自己也为此苦恼过很长一段时间,随着自己技术水平得提高,或多或少地也掌握了ELISA体系的脾气,也是熟能生巧吧,现在做方阵,做ELISA已是轻车熟路了,以前检测一种抗体用整整一下午还算得晕晕的,现在给我十个八个的从包被到显色,一个工作日基本搞定.下面就由我抛砖引玉地来说一下,做ELISA的经验总结:1.包被原的性质很重要,蛋白浓度,是否降解,这关系到你做出的抗体可不可以被其识别,所以保存抗原很重要,我做重组蛋白时,师兄都严格警告我一定要在冰浴下缓慢融化就是这个道理.还有有的包被原可能不是蛋白,对于生物素和脂类物质或小分子物质我们要事先对其改造再加以包被,我把方法简要的列出如下:①亲和素生物素:先亲和素先包被载体,加入生物素化的DNA,这种包被方法均匀、牢固,已扩大应用于各种抗原物质的定量测定。②脂类物质:可将其在有机
实验流程:多靶点免疫荧光染色的实验操作与普通单标记免疫组化流程相似。试剂盒中的荧光染料可以在HRP酶的作用下将信号共价结合到抗原上,随后即可衔接下一轮单标染色,直至全部标记完成,即可复染DAPI后封片观察。1.脱蜡水合:1)新鲜二甲苯浸片10min,重复3次。2)梯度乙醇浸片,100% 5min;95% 5min;70% 2min。3)灭菌水洗片1min,重复3次。4)10%中性福尔马林浸片10min,灭菌水洗片1min,重复3次。2.微波修复:1)将脱蜡水合后的玻片置于修复杯中,用抗原修复液1×工作液浸没。2)将修复杯置于微波炉内高火煮沸。3)低火维持15min。4)取出室温自然冷却至室温。3.封闭:1)去除玻片上残存洗液。2)用组画笔圈出玻片上的样本区域,滴加封闭剂,覆盖样本区域。3)室温保湿震荡10min。4.一抗孵育:1)去除玻片上的封闭剂。2)用移液器滴加稀释的一抗溶液,浸没样本区域。3) 37℃恒温保湿震荡孵育1hr。4)用1×TBST buffer浸洗玻片3min,重复1次。5.二抗孵育:1)去除玻片上残存的洗液。2)直接滴加二抗溶液,浸没样本区域。3)室温保湿震荡孵育1
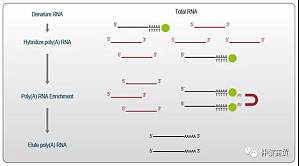
样本RNA提取、去除残留DNA及RNA质检后,我们需要决定是否以及如何去富集感兴趣的RNA。总RNA中含有大量的核糖体 RNA (rRNA),约占总RNA 80 – 98%的比例。对绝大多数 RNA-Seq 应用来说,需要去除 rRNA 或富集mRNA,以将测序资源集中在转录组所需的部分并节省成本。除核糖体 RNA 外,样本还可能含有其他丰富的转录本,例如,血液样本中的珠蛋白 mRNA 可占所有 mRNA 分子的 30 – 80%。如果不除去这些占比很大的globin mRNA,我们测序 ...
关于丁香通
公司信息
个人用户
企业机构


