Quantitative Protein Profiling by Mass Spectrometry Using Label-Free Proteomics
互联网
799
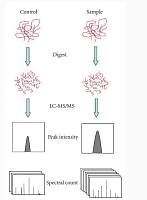
“Gel-free,” or mass spectrometry (MS)-based, proteomics techniques are emerging as the methods of choice for quantitatively comparing proteins levels among biological proteomes, since they are more sensitive and reproducible than two dimensional gel (2-DE)-based methods. Currently, the MS-based methods utilize mainly stable isotope labels (e.g., ICAT�, iTRAQ™) that enable easy identification of differentially expressed proteins in two or more samples. “Label-free” MS-based methods would alleviate several limitations of the labeling methods, provided that relative quantitative profiling of proteins among multiple MS runs is achievable. However, comparisons of multiple MS runs of highly complex biological samples are very challenging and time consuming. To alleviate this problem, several laboratories and MS vendors have developed software for computer-assisted comparisons of multiple label-free MS runs to allow profiling of differentially expressed proteins. In this chapter, we describe the use of custom-developed MatchRx software in quantitative comparison of multiple label-free MS runs. We also describe details of sample preparation, fractionation, statistical analysis, and protein database searching for label-free comparative quantitative proteomics, as well as the application of a “targeted” MS approach, which includes quantification of the samples using MS followed by selective identification of only the differentially expressed peptides using tandem MS (MS/MS).