测序技术及DNA序列测定结果分析
互联网
实验试剂
(1)SILVER SEQUENCETM DNA测序试剂盒。
(3)10%过硫酸铵,0.5g过硫酸铵溶于4ml水中,定容至5ml,应新配新用。
(5)TBE电极缓冲液:10×TBE 缓冲液稀释至1×TBE备用。
(8)染色溶液:硝酸银2克,甲醛3ml,溶于2升超纯水中备用。
(9)显影溶液:60克碳酸钠(Na2CO3)溶于2升超纯水中,使用前加3ml 37% 甲醛和 40ml硫代硫酸钠溶液(10mg/ml)。
(12)Sigmacote (Sigma CAT. #SL-2)。实验步骤
(1) DNA的浓度和纯度必须经过琼脂糖凝胶电泳或荧光法测定, 样品应与已知量DNA一起电泳。
(3) DNA制备过程中用核糖核酸酶处理所产生核糖核苷酸,虽然它们在电泳后DNA样品的前面,并不能观察到,但它们仍会有260nm光吸收。
(一)测序反应:
2. 对于每组四个测序反应,在一个eppendorf管中混合以下试剂:
3. 在引物/模板混合物(以上第2步)中加入1.0ml测序级Taq DNA聚合酶(5u/ml)。用吸液器吸动几次混匀。
4. 从第3步的酶/引物/模板混合物中吸取4ml加入每一个d/ddNTP混合物的管内。
5. 在微量离心机中离心一下,使所有的溶液位于eppendorf管底部。
6. 把反应管放入预热至95℃的热循环仪,以[注意]中循环模式为基准,开始循环程序。对于每个引物/模板组合都必须选择最佳退火温度。下列程序一般能读出从引物开始350碱基的长度。
7. 热循环程序完成后,在每个小管内加入3μl DNA测序终止溶液,在微量离心机中略一旋转,终止反应。
(二)、测序凝胶板的制备
A. 在1ml 95%乙醇, 0.5%冰乙酸中加入5ml粘合硅烷(Bind Silane), 配成新鲜的粘合溶液。
B. 用经浸透新配的粘合溶液浸透的吸水棉纸擦拭仔细清洗过并已经自然干燥的玻璃板, 整个板面都必须擦拭。
C. 4-5分钟后, 用95%乙醇单向擦玻璃板, 然后略用力沿垂直方向擦拭。重复三次这一清洗过程, 每次均须换用干净的纸, 除去多余的粘合溶液。
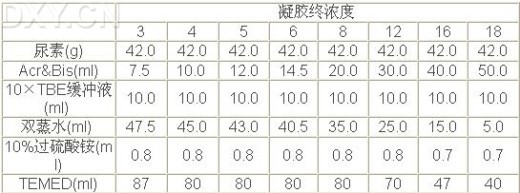
(3)胶配制好后,即可灌制胶板。一般是将凝胶沿着压条边缘缓慢地倒入玻璃板的槽中,倒完后,静止放置使之聚合完全。
(三)电泳:
(1)当凝胶聚合完全后,拨出鲨鱼齿梳,将该梳子反过来,把有齿的一头插入凝胶中,形成加样孔。
(2)立即将胶板固定在测序凝胶槽中,一般测序凝胶槽的上下槽是分开的,因而只有在固定好凝胶板后,方能加入TBE缓冲液。
(3)稀释10×TBE缓冲液至1×TBE,将该缓冲液加入上下二个电泳槽中,去除产生的气泡,接上电源准备预电泳。
(5)按30V/cm的电压预电泳20-30分钟。预电泳的过程是去除凝胶的杂质离子,同时使凝胶板达到所需的温度。高温电泳可防止GC丰富区形成的发夹状结构,影响测序的结果。
染色过程要求凝胶浸在塑料盘中。因而至少使用两个盘子,大小与玻璃板类似。在盘中加入新鲜溶液之前须用高质量的水洗涤盘子。
1. 电泳完毕后用一个塑料片子小心地分开两板,凝胶应该牢固地附着在短玻璃板上。
2. 固定凝胶:将凝胶(连玻璃板)放入塑料盘,用固定/停止溶液浸没,充分振荡20分钟或直至样品中染料完全消失,胶可在固定/停止溶液中保存过夜(不振荡)。保留固定/停止溶液,用于终止显影反应。
3. 洗胶:用超纯水振荡洗胶3次,每次2分钟。从水中取出, 当转移至下一溶液时拿着胶板边沿静止10-20秒,使水流尽。
(1). 在显影溶液中加入甲醛(3ml)和硫代硫酸钠溶液(400μl)以完成显影液的配制。
(3). 立刻将凝胶转移至1升(总量的一半)预冷的显影液充分振荡直至模板带开始显现或开始出现第一批条带,把凝胶移入剩下的1升显影液中继续显影2--3分钟,或直至所有条带出现。
6. 固定凝胶:在显影液中直接加入等体积的固定/停止溶液。停止显影反应,固定凝胶。
7. 在超纯水中浸洗凝胶两次,每次2分钟,注意在本操作中戴手套拿着胶板边缘避免在胶上印上指纹。
8. 将凝胶置于室温干燥或用抽气加热法干燥。在可见光灯箱或亮白,黄色背景(如纸)上观察凝胶,若需永久保存的记录, 则可用EDF胶片保留实验结果。
(五)、EDF胶片显影
使用EDF胶片可增强测序条带的对比度,如果测序胶上条带很淡,我们建议把数据转移至EDF胶片,银染胶在其影像转移至EDF胶片之后可增强条带可读性。
2. 在红灯下找到EDF胶片有缺刻的一角, 然后把胶片置于凝胶上,使缺口位于左上角。由于EDF胶片是单面的,因此必须确保缺口在左上角。
3. 在EDF胶片上放置一干净、干燥的玻璃板,开亮灯箱约20秒。
4. 用冲显放射自显影胶片的步骤手工显影EDF胶片,可使用下列操作过程:
d. 水洗1分钟.注意事项
由于超螺旋质粒产生的信号比松驰的线性双链DNA弱,因此使用超螺旋质粒作为模板时其用量要比其它模板大一些。
2、计算与4.5pmol相当的引物纳克数可用以下一般公式:
dsDNA:1pmol=(6.6×10-4 mg)×n,其中n为模板碱基对数
95℃ 2分钟。然后: 95℃ 30秒(变性), 42℃ 30秒(退火), 70℃ 1分钟(延伸)。