

计算流式细胞术:高维算法深度挖掘多色流式数据

以Cytek FSPTM为代表的全光谱流式检测技术因其强大的相似染料分辨能力与自发荧光提取功能,可以在一管样本中完成超过40色的同时检测,引领流式进入高维时代。超多色流式数据中包含大量的生物学信息,往往很难通过传统的手动圈门充分展示。近年来,计算流式细胞术高速发展,多种降维、聚类、预测建模算法为高维流式分析提供了强大的解决方案。本期推文将综述高维流式数据解析流程,讨论如何选择合适的算法进行数据深度挖掘。
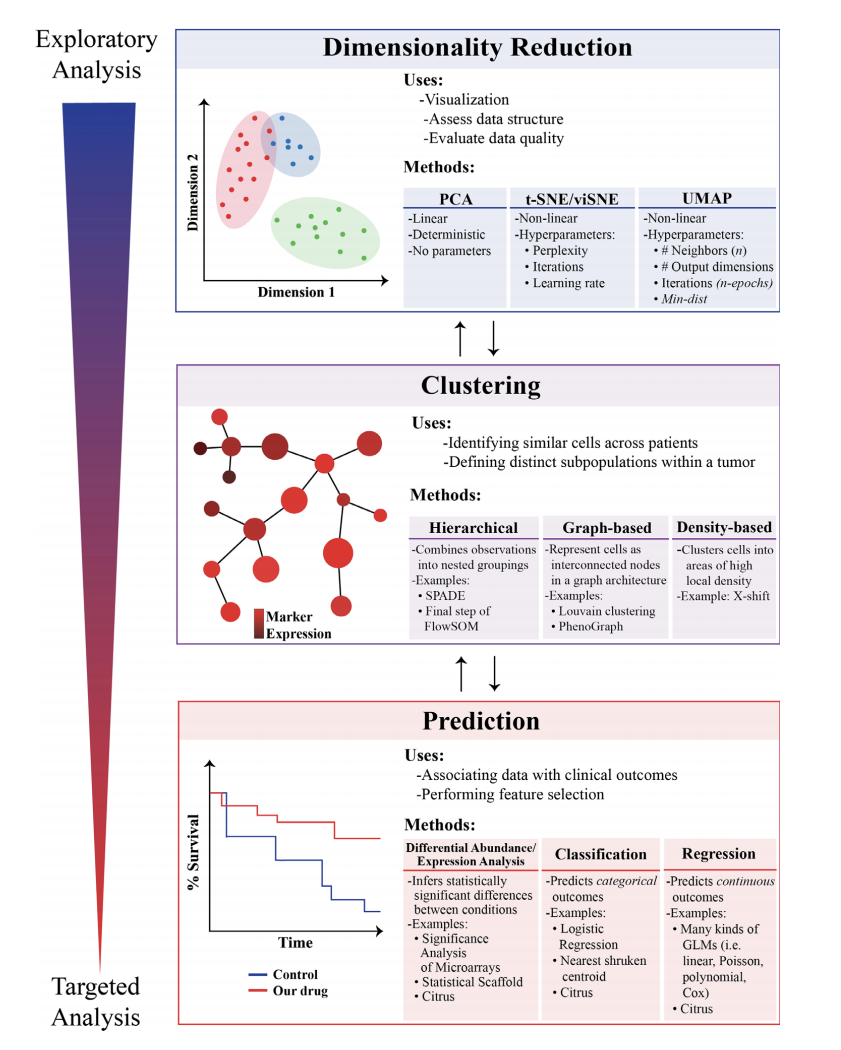
图1. 高维流式数据分析流程
· 高维流式数据分析流程 ·
面对高维流式数据的复杂性,数据分析策略通常由更具探索性的降维可视化算法开始,进行至无监督聚类自动圈门,最终通过预测建模深度挖掘样本生物学相关信息(也有观点将数据前处理算法纳入讨论,如使用flowAI/flowClean等算法完成,本文中不做重点介绍)。考虑到数据处理流程的整体性,每个阶段的分析均会对其他阶段产生影响,上游的数据分析结果对于下游算法的选择具有指导意义,反之亦然。
● ● ● ● ● ● ●
数据降维
数据降维是通过无监督机器学习算法,仅基于每个数据点的测量值,在二维或三维空间对单细胞数据可视化的常用方法,可以揭示细胞在高维空间的分布趋势,也可以用来评估多个实验或数据集的数据质量。常用的降维算法包括主成分分析(PCA,线性降维)、t分布随机邻位嵌入(t-SNE,非线性降维)以及一致流行逼近与投影(UMAP,非线性降维)算法(图2)。由于生物学数据中充斥着多种信号放大、正负反馈等非线性过程,存在多项式、指数或其他高度复杂的关系,两种非线性降维算法(t-SNE, UMAP)能够更好地捕捉数据局部信息,PCA则常用作非线性降维的预降维手段用于提升t-SNE与UMAP降维质量。1
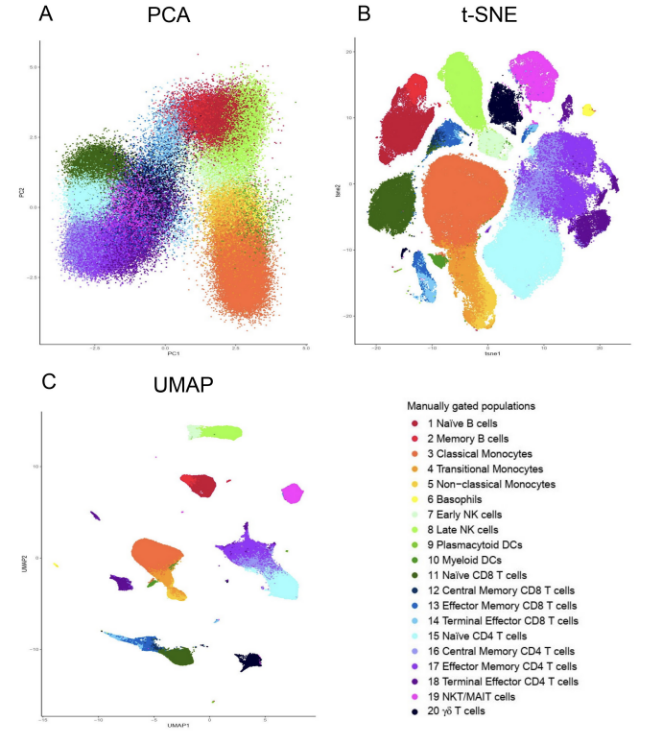
图2. PCA、t-SNE及UMAP降维结果展示
t-SNE是高维流式分析中最常用的降维手段,通过对高维空间中所有点进行两两比较,根据高斯分布将点与点之间的距离(欧氏距离)转换为相邻概率矩阵;随后将所有数据点随机分布于低维空间(通常为二维),通过t分布建立低维空间中数据点的相邻概率矩阵;最后进行迭代以最小化两个矩阵的差异,从而优化散点(细胞)在二维空间中的位置分布,在降维数据中保留高维空间信息。结果展示中,相似的散点会彼此靠近,不同的细胞亚群得以直观展现。2
在使用t-SNE分析数据前,需设置如迭代次数(Iterations)、困惑度(Perplexity)等超参数,超参数设置将对最终结果产生显著影响(图3)。其中困惑度反映降维对原始数据局部与全局信息之间的平衡,通常在5-50间调整,如困惑度较低时算法认为每个数据点仅有少量的临近点,此时输出数据优先保持原始数据局部信息。迭代次数的调整意味着更优降维结果与运算时间的平衡。需注意的是,t-SNE算法无法保留原始数据的密度与全局距离,这意味着t-SNE降维结果中较远类群间的距离无法代表其相似性,降维后类群大小也无法对应其在高维空间中的大小。此外由于t-SNE的随机性,多次处理相同数据时将产生不同的降维结果。
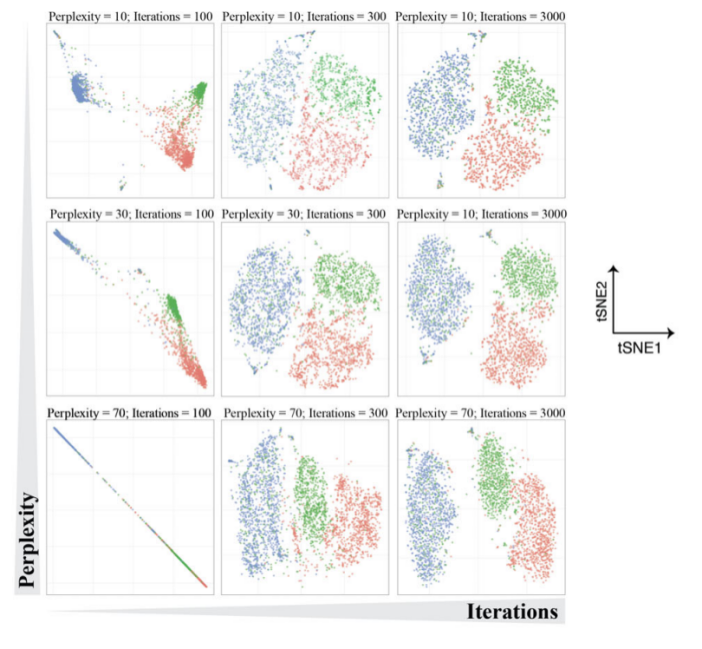
图3. t-SNE不同超参数设置对降维结果的影响
UMAP的降维流程与t-SNE是类似的,但在高维空间相邻数据点的判定中利用了流形理论和黎曼几何相关算法。算法的改进使得UMAP具有更快的运行速度,并可以在降维数据中更好保留原始数据的全局特征(图4)。在进行数据降维前,同样需要预先优化几个超参数,分别包括n(认为某一数据点周围最近的n个点为邻居)、d(数据输出所嵌入的维度)、n-epoch(迭代次数)、min-dist(降维数据中点与点之间的最小距离)。其中n的设定最为重要,更大的n值将能够更好地保留数据的全局结构;min-dist为美学参数,数值越大表示细胞群之间更加分散。在使用时,可以固定d和n-epoch,并测试不同的n、min-dist,以达到最佳的降维表现。1,3

图4. 对猛犸象三维数据点进行降维时,
相比于t-SNE,UMAP能够更好地展现数据全局特征
● ● ● ● ● ● ●
聚类分析
降维算法在探索性数据分析的可视化中起到不可替代的作用,但无法明确区分样本中细胞亚群的组成结构。因此,高维流式数据分析的第二阶段通常使用聚类算法,根据样本中细胞的相似性,自动、无偏倚地将数据划分为可量化的子集。据不完全统计,现有聚类方法超过30种,使用不同聚类方法处理流式数据可产生差异化的计算结果(图5)。4免疫学分析中,SPADE、FlowSOM与PhenoGraph三种聚类算法最为常用,下文将主要对这三种无监督聚类方式进行介绍与对比。
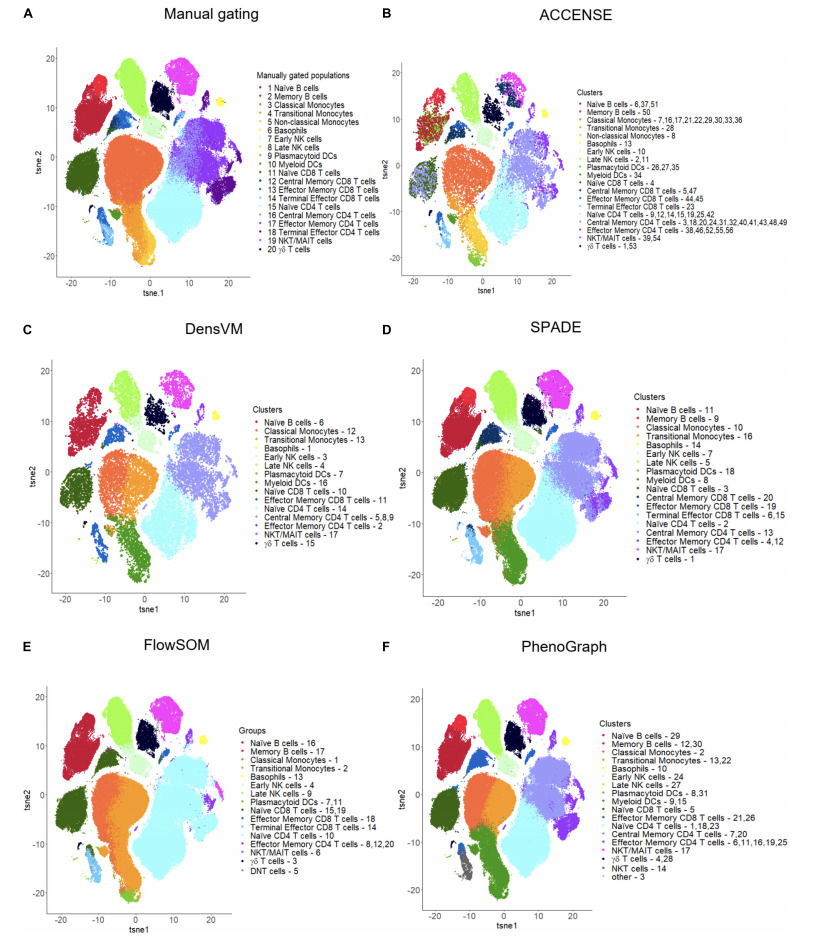
图5. 手动圈门与不同聚类算法分群效果对比
三种聚类方法中,SPADE与FlowSOM均属于层次聚类(Hierarchical clustering)
,层次聚类是生物信息学中最常用的聚类方式,通过将数据中的每个数据单元组合为更大类群(Bottom-up),或将所有数据作为整体集群逐渐划分为更小的类群(Top-down)完成聚类,并以迭代的方式缩小类群数量,直至达到用户指定的类群数量。1
SPADE(Spanning tree progression of density normalized events)是高维流式分析中第一个被广泛运用的聚类手段,其工作流程包括四步:a. 根据细胞密度对数据进行降采样(Downsample),以均衡稀有和高丰度细胞群的权重;b. 通过自下而上的方式进行聚类,并将表型相似的细胞划分为簇或节点;c. 在所有的节点之间构造最小生成树(Minimun spanning tree,MST);d. 通过上采样(Upsample)将数据中所有细胞映射到MST的每个簇中。SPADE算法中降采样步骤可以在一定程度平衡高丰度与稀有细胞在聚类中的权重问题,MST数据展示方式也可直观展现各类群之间的层次关系,这使得SPADE在计算流式细胞术发展初期受到广泛关注,但SPADE的数据展示方式无法在每个簇/节点中反映多个标志物的表达情况(图6.A),且较慢的数据处理速度无法很好兼容细胞数目过大的流式数据。5
相比之下,2015年Van Gassen提出的FlowSOM算法可通过构建自组织映射(Self-organizing maps)与一致性层次聚类快速完成大量数据的聚类分析,并通过MST或热图(Heatmap)输出聚类结果。该算法在满足SPADE类似功能的前提下大幅提高了运算速度,无需降采样步骤,仅12秒即可完成180万个细胞的聚类分析;优化的可视化方式使研究人员能够在一张星状图中清晰展现各簇的标志物表达情况(图6.B)。6 FlowSOM可以通过增加预设分群数量提升发现稀有细胞类群的可能,但过高的预设分群数量可能会引起高丰度细胞类群的分裂,并降低数据重现性。
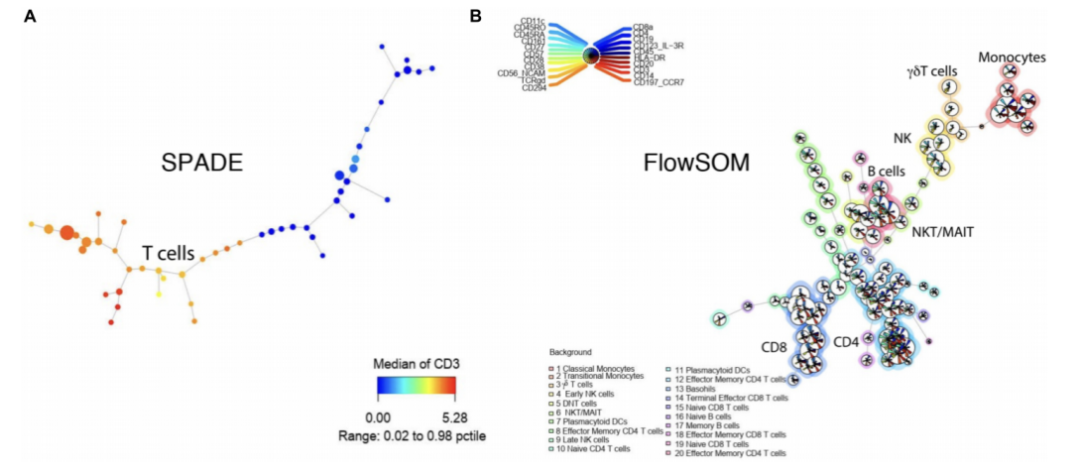
图6. SPADE与FlowSOM聚类可视化数据对比
第三种常用的降维算法,PhenoGraph,属于图聚类(Graph-based clustering)自动分群算法。利用点(代表每个细胞)与边(代表细胞间的邻居关系)构建网络,并通过Louvain聚类(通过迭代使输出数据模块度最高的聚类方式)对表型相似的细胞进行分群(图7)。得益于Louvain聚类的算法优势,PhenoGraph可以对丰度仅为0.05%的稀有细胞进行自动分群,聚类结果与手动圈门具有较强可比性。该方法只需定义超参数k(构建原始网络时每个细胞最近的邻居数量)即可进行聚类,无需进一步指定目标分群数量。7
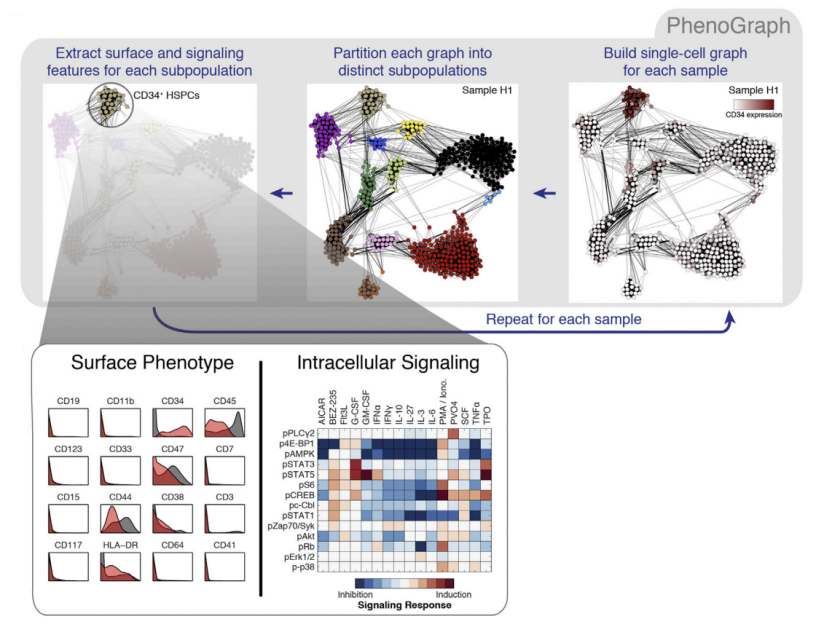
图7. PhenoGraph数据处理流程
考虑到以上三种聚类方式的特点,在针对具体数据分析时可根据数据情况匹配更适合的算法。如数据量较大且计算时间很重要时,应优先考虑FlowSOM;当希望获取细胞类群的层次结构与表型关系时,可选用SPADE或FlowSOM层次聚类算法;若数据中包含稀有细胞亚群或不确定数据中的类群数量时,可选用PhenoGraph进行分析。1
● ● ● ● ● ● ●
预测模型
通过上述降维、聚类工具的组合,已能够将具有不同标志物表达特征的细胞分群并进行可视化展示,但在细胞类群被确定后,我们仍需要利用预测建模等统计学技术寻找细胞类群之间某些指标(如丰度、标志物表达、临床或实验条件等)的关联与差异,以合理解释数据。例如Citrus及Statistical Scaffold算法常被用于细胞丰度与标志物表达的差异分析;Wanderlust、Wishbone与Monocle算法被广泛用于细胞发育建模。1,6,8 由于该部分内容信息量较大,我们将在后期推文中进行专题介绍,欢迎各位读者关注Cytek微信公众号,跟踪光谱流式使用技巧与前沿进展。
● ● ● ● ● ● ●
总结
>>
计算流式细胞术经过十余年的发展,在算法层面已较为成熟,多种算法作为全新的工具集,为高维流式数据的可视化、解释与建模开辟了全新途径。虽然一些商品化流式分析软件支持部分算法的图形界面处理,更多算法的灵活组合仍需通过编程框架(如R或MatLab)中的开源包使用。本期推文为大家介绍了计算流式细胞术分析流程并重点讨论了常用的降维、聚类算法的工作原理与性能评估,更多算法相关信息欢迎阅读参考文献原文。
参考文献:
1.Keyes T J, Domizi P, Lo Y C, et al. A cancer biologist's primer on machine learning applications in high‐dimensional cytometry[J]. Cytometry Part A, 2020, 97(8): 782-799.
2.Van der Maaten L, Hinton G. Visualizing data using t-SNE[J]. Journal of machine learning research, 2008, 9(11).
3.https://pair-code.github.io/understanding-umap/
4.Liu P, Liu S, Fang Y, et al. Recent advances in computer-assisted algorithms for cell subtype identification of cytometry data[J]. Frontiers in cell and developmental biology, 2020, 8: 234.
5.Qiu P, Simonds E F, Bendall S C, et al. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE[J]. Nature biotechnology, 2011, 29(10): 886-891.
6.Rybakowska P, Alarcón-Riquelme M E, Marañón C. Key steps and methods in the experimental design and data analysis of highly multi-parametric flow and mass cytometry[J]. Computational and Structural Biotechnology Journal, 2020, 18: 874-886.
7.Levine J H, Simonds E F, Bendall S C, et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis[J]. Cell, 2015, 162(1): 184-197.
8.Saeys Y, Van Gassen S, Lambrecht B N. Computational flow cytometry: helping to make sense of high-dimensional immunology data[J]. Nature Reviews Immunology, 2016, 16(7): 449-462.
关于Cytek
About Cytek /
Cytek® Biosciences, Inc.(Nasdaq: CTKB)作为一家全球生命科学技术公司,通过其受专利保护的全光谱分析(Full Spectrum Profiling,FSP™)技术,提供高分辨率、高参数和高灵敏度的新一代细胞分析工具。Cytek的创新技术通过检测荧光信号的完整光谱信息,以实现更高水平更高灵敏度的多参数检测。Cytek的FSP™平台包括其核心仪器—Aurora和Northern Lights™分析系统、Aurora CS分选系统、试剂、软件和服务,为客户提供全面和完整的解决方案。Cytek总部位于美国加利福尼亚州Fremont,在全球设有分部和分销渠道。
更多的相关信息,请登录Cytek的官方网站:www.cytekbio.com和www.cytekbio.com.cn。
注:Cytek®, Tonbo Biosciences, cFluor®, Full Spectrum Profiling™, FSP™和Northern Lights™是Cytek Biosciences, Inc. 的商标或注册商标。Cytek®全光谱检测技术相关专利包括但不限于:US10739245B2,US11169076B2,US10788411B2。
/


本周
本月
本年
- 促销公告
- 更多 ›
你可能感兴趣的产品
- 实验法 vs 计算法:蛋白互作检测的优缺点对比
- 人 ATOX1 AssayLite 多色偶联抗体流式细胞术试剂盒
- TCGA数据挖掘服务| 肿瘤相关表观遗传调控基因深度分析
- 人着丝粒蛋白B(CENP-B)多色结合抗体流式细胞术试剂盒|Human Centromere Protein B (CENP-B) Multi-Color Conjugated Antibodies Flow Cytometry Kit
- FyMeso人间充质干细胞无血清培养基(chemical defined)
- 冷冻研磨仪/低温冷冻研磨仪/组织研磨仪
- 美国PE耗材配件珀金埃尔默GC色谱耗材
- 外泌体提取纯化试剂盒(组织、细胞上清、血液、尿液等)
- RNA快速提取试剂盒(带DNA清除柱 )
- BVC professional 真空吸液系统(废液处理器)
- 链酶蛋白酶浓缩液(20X)
- 氘灯Waters 486 D2 lamp 2000时
