IF 44.5!中山大学结直肠癌领域发布最新成果
2025-11-15 18:00点击次数:77
关键词:KRAS 基因是结直肠癌发生发展的关键驱动基因,约 40% 的结直肠癌患者携带 KRAS 突变。长期以来,这种突变型结直肠癌缺乏有效疗法,患者预后较差。KRAS G12C 抑制剂(如 sotorasib 和 adagrasib)联合 EGFR 抗体的应用显著改善了 KRAS G12C 突变结直肠癌的治疗效果,客观缓解率达 34%–46%,获得 FDA 加速批准,为患者带来新的生存希望【1-2】。
然而,几乎所有患者最终都会出现耐药,成为当前临床主要挑战。目前已发现多种导致 KRAS/EGFR 双靶治疗耐药的遗传学机制,如继发性 KRAS 突变、RAS 上下游信号或旁路再激活等,但约 1/3 复发病例未检测到基因变化,提示非遗传机制同样关键【3-4】。
近年来,肿瘤细胞谱系可塑性作为非遗传耐药的重要机制备受关注。在 KRAS 突变型非小细胞肺癌中,不同研究团队报道了肺腺癌细胞在 KRAS 靶向治疗下可转分化为一型肺泡上皮样(AT1)、鳞状(squamous)或黏液样(mucinous)细胞等不同耐受状态;在胰腺癌中,KRAS 抑制剂可诱导肿瘤细胞向经典型(Classical)状态转变【5-8】。这些发现表明,肿瘤细胞能通过多种不同的转分化路径进入耐药状态以逃避药物压力。结直肠上皮细胞本身具有高度可塑性,可在应激或损伤条件下去分化为干细胞样状态以完成组织的再生修复。既往研究发现,结直肠癌在化疗压力下可转变为胎儿前体样(fetal progenitor)状态并进一步分化为鳞状(squamous)或神经内分泌谱系(neuroendocrine-like state)【9】。那么,在 KRAS/EGFR 双靶治疗压力下,结直肠癌细胞是否会发生特定谱系转变? 这种谱系转变是否促进耐药?背后的分子机制是什么?能否通过干预谱系转变的核心机制来延缓耐药?
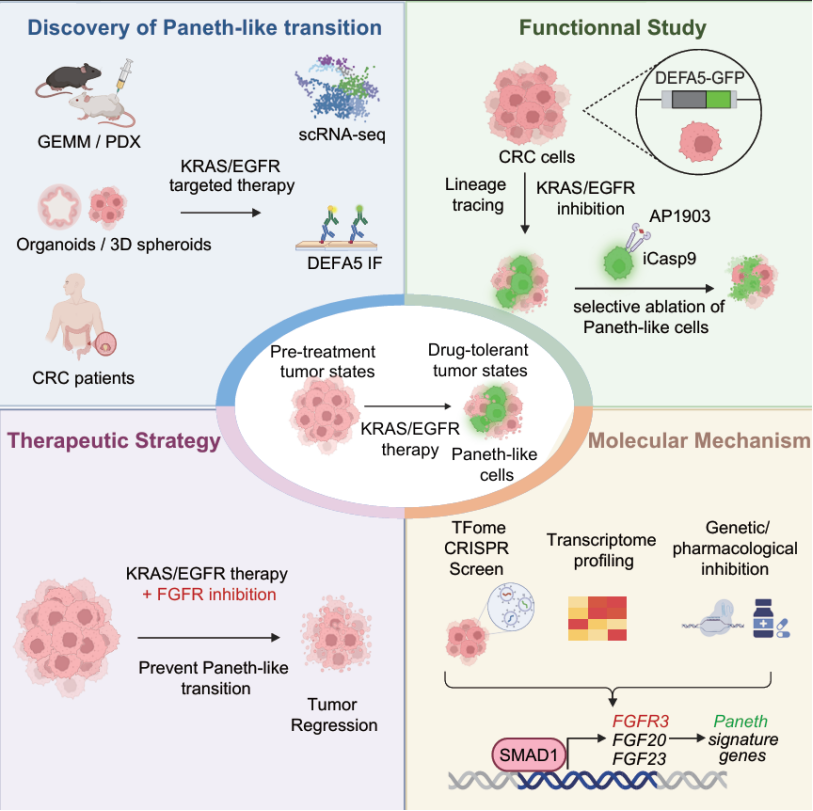
2025 年 11 月 13 日,中山大学肿瘤防治中心高益军、王峰、廖雯婷、赵齐与方兆元团队合作,在 Cancer Cell 发表题为 Paneth-like transition drives resistance to dual targeting of KRAS and EGFR in colorectal cancer 的研究论文。本研究利用 KRAS 突变型结直肠癌类器官(PDO)、患者来源异种移植瘤(PDX)及基因工程小鼠模型(GEMM),结合单细胞转录组测序与谱系示踪技术,系统解析了 KRAS/EGFR 双靶治疗下的细胞命运变化。研究发现,结直肠癌细胞在治疗压力下发生向类潘氏细胞(Paneth-like)状态的谱系转变,并进一步阐明了转录因子 SMAD1 通过激活 FGFR3 信号通路促进类潘氏转分化、驱动治疗耐药的分子机制,为靶向肿瘤谱系可塑性延缓耐药提供了全新思路。

研究团队在 iKAP(tet-O-LSL-KrasG12D, ApcL/L, Trp53L/L, VillinCreERT2)基因工程小鼠模型中,诱导结直肠癌后给予 KRAS/EGFR 双靶治疗,发现残留病灶中潘氏细胞特征显著富集。潘氏细胞是一类位于小肠隐窝底部的分泌型上皮细胞,通过分泌 Wnt3a 等生长因子支持肠道干细胞功能,并分泌抗菌肽维持肠道免疫屏障。团队进一步在 iKAP 肿瘤、CRC 细胞系 3D 球状体及 PDO 模型中均观察到潘氏细胞标志基因(DEFA5、DEFA6)在治疗后显著上调。单细胞转录组分析显示,具有潘氏特征的细胞比例随治疗进程持续上升,轨迹分析提示 CRC 细胞经由滞育样耐药持久细胞(Diapause-like drug-tolerant persister)中间态逐步转变为类潘氏状态。
为动态追踪这一谱系转变过程,研究团队在 CRC 细胞系中构建了 DEFA5 启动子驱动的 Cre 重组酶示踪系统,发现 KRAS/EGFR 治疗后 DEFA5 阳性细胞比例逐步上升。通过在内源性 DEFA5 位点敲入 CopGFP 报告基因,实现了对类潘氏状态的实时示踪,观察到联合治疗 3 天后 CopGFP 阴性细胞开始转变为 CopGFP 阳性类潘氏细胞。其中,类潘氏细胞的比例随治疗时间延长而增加,且在撤药后迅速下降,表明类潘氏状态转分化是药物直接诱导的适应性反应。进一步利用 iCasp9 系统定向清除类潘氏细胞后,显著增强了肿瘤对双靶治疗的敏感性,证实类潘氏细胞在耐药复发中的核心作用。
为解析类潘氏细胞转分化驱动 KRAS/EGFR 靶向治疗耐药的分子机制,研究团队基于 CRC 细胞 3D 球状体培养体系开展了转录因子文库的 CRISPR/Cas9 功能基因组筛选,并结合转录因子与潘氏细胞标志基因集的表达相关性分析,鉴定出 SMAD1 是驱动类潘氏转分化引起耐药的关键转录因子。在体内外模型中敲除 SMAD1 均能显著阻断肠癌细胞转分化并逆转耐药。进一步的机制研究揭示 SMAD1 通过结合到 FGFR3 的启动子区,上调其转录,从而激活 FGFR3 下游信号通路,促进肿瘤细胞向类潘氏状态转变;FGFR3 持续激活进一步引发类潘氏细胞中 MAPK 信号重新活化,导致药物耐受。遗传学敲除或药物抑制 FGFR3 在 CRC 3D 球状体、iKAP 及 PDX 模型中均可阻断类潘氏转分化并增强肠癌对于 KRAS/EGFR 联合治疗的敏感性,提示 FGFR3 信号是谱系可塑性驱动耐药的关键节点。为验证临床相关性,研究团队分析了两例 KRAS G12C 突变 CRC 患者在 KRAS/EGFR 治疗前后的配对活检样本,发现治疗后 DEFA5 阳性类潘氏细胞显著富集,从患者样本层面证实了类潘氏转分化介导的耐药机制在 KRAS 突变结直肠癌中的普适性。
本研究揭示了结直肠癌细胞谱系可塑性在 KRAS/EGFR 靶向治疗耐药中的核心作用,阐明了转录因子 SMAD1 通过激活 FGFR3 信号通路诱导肠癌细胞向类潘氏状态转分化,从而驱动双靶治疗耐药的分子机制。本研究深化了对肿瘤细胞命运重塑与治疗耐受关系的理解,并提出联合抑制 FGFR3 作为延缓耐药的新策略,为克服 KRAS 突变型结直肠癌中可塑性驱动的治疗耐受指明了新方向。
中山大学肿瘤防治中心高益军研究员、王峰教授、廖雯婷研究员、赵齐研究员为本文的共同通讯作者;博士生张跃桐、硕士生陈嘉颖、硕士生佘勇、浙江大学方兆元研究员为共同第一作者。本研究得到纪念斯隆凯特琳癌症中心 Rona Yaeger、中山大学肿瘤防治中心王自峰、武远众、邓蓉、王瑞萍、华中农业大学颜彦、中国科学院动物所吕赫喆、西湖大学季红斌、浙江大学冯新华、生化细胞所高栋、中山大学邝栋明、华中科技大学孙书国的大力支持与帮助。
招聘启事
中山大学肿瘤防治中心、华南恶性肿瘤防治全国重点实验室高益军团队长期聚焦肺癌、结直肠癌对 RAS/MAPK 通路靶向治疗的耐受机制。团队通过构建肿瘤类器官、PDX 及小鼠自发肿瘤等多层级疾病模型,结合多组学分析与功能基因组筛选,解析肿瘤谱系可塑性与微环境重塑在治疗耐受中的作用,探索逆转耐药的新策略。课题组负责人高益军研究员获国家海外高层次人才引进计划、国家创新研究群体项目青年项目、国家重点研发计划青年科学家项目、国自然面上等项目资助,作为通讯作者或第一作者的论文发表于 Cancer Cell、Nature Medicine、Cancer Discovery(2 篇)、Cell Research、Signal Transduction and Targeted Therapy 等国际高水平期刊。现拟招聘博士后 2–3 名,具有生物信息学、表观遗传或肿瘤免疫研究背景者优先。实验室将提供优越的科研平台与具有竞争力的薪资待遇,支持申报各类科研基金(博新计划、博士后基金、国自然青年基金等)。
申请方式:请将个人简历及相关材料发送至 gaoyj@sysucc.org.cn
原文链接:
https://www.cell.com/cancer-cell/fulltext/S1535-6108(25)00451-9
如需代发文章宣传、新闻稿、招聘等,请后台回复【学术】添加小编
我们长期为科研用户提供前沿资讯、实验方法、选品推荐等服务,并且组建了 70 多个不同领域的专业交流群,覆盖神经科学、肿瘤免疫、基因编辑、外泌体、类器官等领域,定期分享实验干货、文献解读等活动。
添加实验菌企微,回复【】中的序号,即可领取对应的资料包哦~
【2401】论文写作干货资料(100 页)
【2402】国内重点实验室分子生物学实验方法汇总(60 页)
【2403】2024 最新最全影响因子(20000+ 期刊目录)
【2404】免疫学信号通路手册
【2405】PCR 实验 protocol 汇总
【2406】免疫荧光实验 protocol 合集
【2407】细胞培养手册
【2408】蛋白纯化实验手册
【2501】染色体分析方法汇总
【2502】国自然中标标书模板
【2503】WB 实验详解及常见问题解答
【2504】DeepSeek 论文写作常用口令
【2505】中国科学院期刊分区表(2025 年最新版)
【2506】期刊影响因子(2025 年最新版)
【2507】130 种实验室常用试剂配制方法(附全套资料)
【2508】常见信号通路
【2509】限制性核酸内切酶大全