

连发两篇 | 突破实体瘤 CAR-T 耐药困局
2025-10-26 18:00点击次数:182
关键词:Carl H. June 教授团队在 2025 年 9 月 30 日于 Nature Reviews Drug 发表综述阐述体内 CAR-T 突破性进展。
仅两周后,Carl H. June 教授团队于 10 月 14 日再于 Nature Reviews Immunology 发表重磅综述「Microenvironmental regulation of solid tumour resistance to CAR T cell therapy」,直指阻碍疗效的核心瓶颈 —— 肿瘤微环境(TME)构筑的三重免疫屏障,深入解析 TME 通过物理封锁、免疫抑制与代谢绞杀瓦解 CAR-T 疗效的机制,并前瞻破局路径。

CAR-T 疗法在在实体瘤治疗中仍面临严峻挑战。
实体瘤通过异常脉管系统和致密细胞外基质(ECM)构筑物理屏障,阻碍 CAR-T 细胞浸润;
突破该屏障的少量细胞继而遭遇免疫屏障压制 —— 既受调节性 T 细胞(Tregs)、髓系抑制细胞(MDSCs)的数量压制,亦因 PD-L1/CTLA-4 等检查点分子上调而功能耗竭。
同时,TME 启动代谢屏障:通过缺氧/营养剥夺削弱细胞活力,并积累乳酸等毒性代谢物。
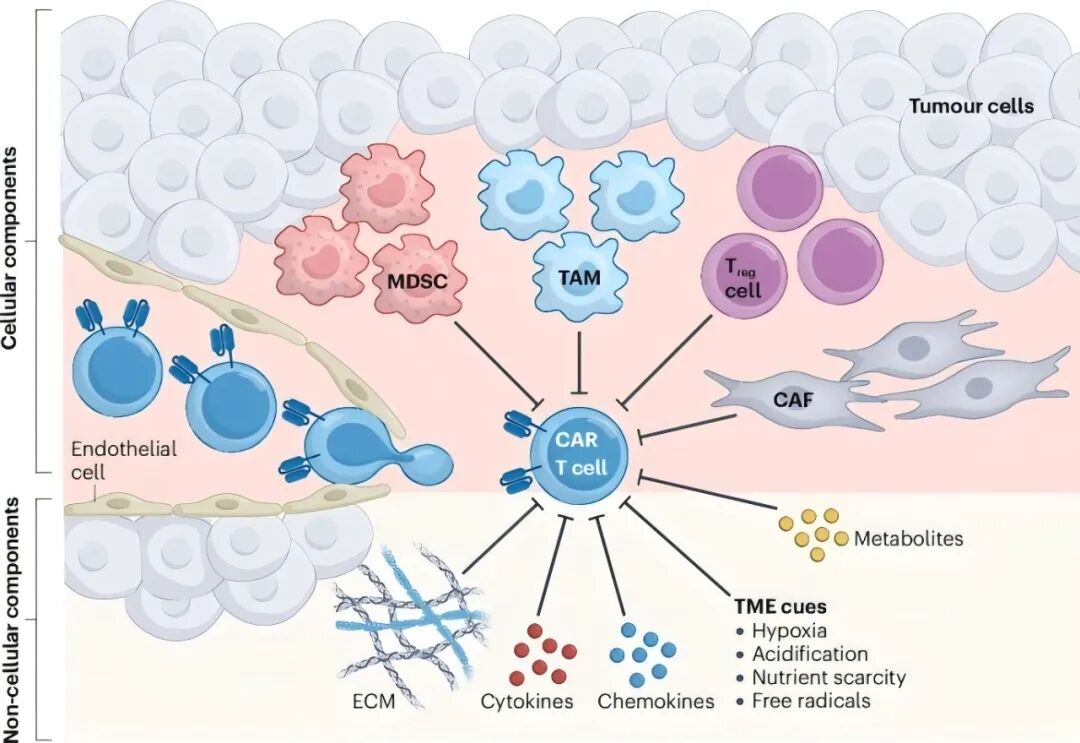
图 1:TME 的细胞和非细胞成分诱导肿瘤对 CAR-T 细胞疗法的耐药性。
因此,突破耐药需三重策略:
抗血管药物与工程化趋化因子受体破解物理屏障;
检查点抑制剂联合抑制细胞清除瓦解免疫屏障;
代谢改造及营养干预对抗代谢屏障。
一、肿瘤脉管系统
肿瘤脉管系统构成 CAR-T 细胞浸润实体瘤的首道屏障。
1、促血管生成因子(如 VEGF)过度表达导致血管结构异常,并显著下调黏附分子,直接阻碍 CAR-T 细胞的外渗过程。
2、内皮细胞(EC)还通过遗传/表观遗传重塑获得免疫抑制特性:一方面抑制黏附分子的表达,另一方面分泌 IL-6、PGE2 等抑制性因子,进而在血管周围构筑由髓系细胞(主导的免疫抑制网络。更致命的是,肿瘤内部的缺氧微环境迫使 EC 依赖糖酵解供能,其代谢产物乳酸及 2-羟基戊二酸 (2-HG) 在局部累积,进一步强化了免疫抑制效应。
这些机制协同作用,导致大量 CAR-T 细胞在试图穿越血管壁的阶段即被拦截并耗竭。
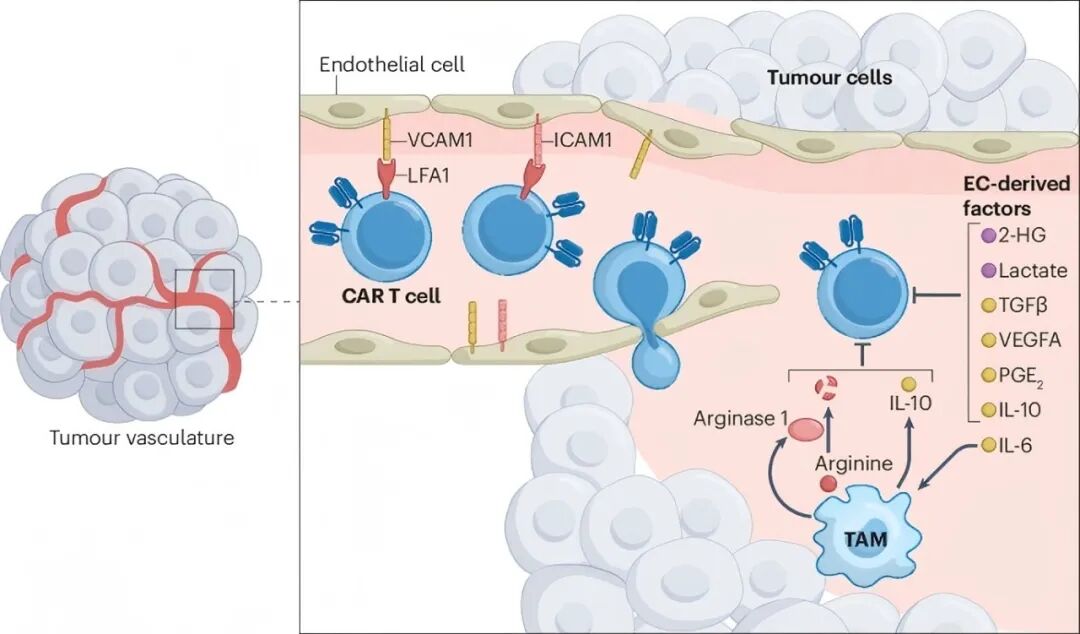
图 2:肿瘤脉管系统抑制 CAR T 细胞活性。
针对此屏障,血管正常化成为核心策略:
低剂量抗 VEGF/VEGFR2 抗体可修复血管结构与功能
同时,靶向 EC 可塑性能恢复黏附分子表达,促进 T 细胞外渗;
另一关键点在于代谢重编程,可逆转 EC 的代谢异常并增强 CAR-T 活性。
二、肿瘤细胞外基质
肿瘤细胞外基质(ECM)的异常重塑构成 CAR-T 细胞浸润实体瘤的第二重屏障。究其本质,癌症相关成纤维细胞(CAF)等基质细胞过度分泌胶原蛋白、透明质酸等成分,形成致密纤维化网络:
一方面通过物理阻塞(<2μm 孔径阻断 T 细胞迁移)直接阻碍 CAR-T 浸润;
另一方面通过诱导 T 细胞功能障碍、破坏趋化梯度及极化免疫抑制性 M2 型巨噬细胞间接削弱抗肿瘤免疫。更关键的是,ECM 硬化挤压脉管系统引发缺氧酸化,进一步抑制 T 细胞活性。
为突破此屏障,ECM 重塑策略聚焦三大路径:
酶解降解:基质金属蛋白酶(MMP)及胶原酶可分解 ECM 组分,但广谱 MMP 抑制剂临床疗效不佳;
靶向交联:抑制赖氨酰氧化酶(LOX)阻断胶原交联,或阻断胶原受体 DDR1 破坏纤维排列,临床前研究证实可提升 T 细胞浸润及免疫疗效;
载体递送:溶瘤病毒搭载胶原酶实现靶向降解,增强 CAR-T 肿瘤募集。
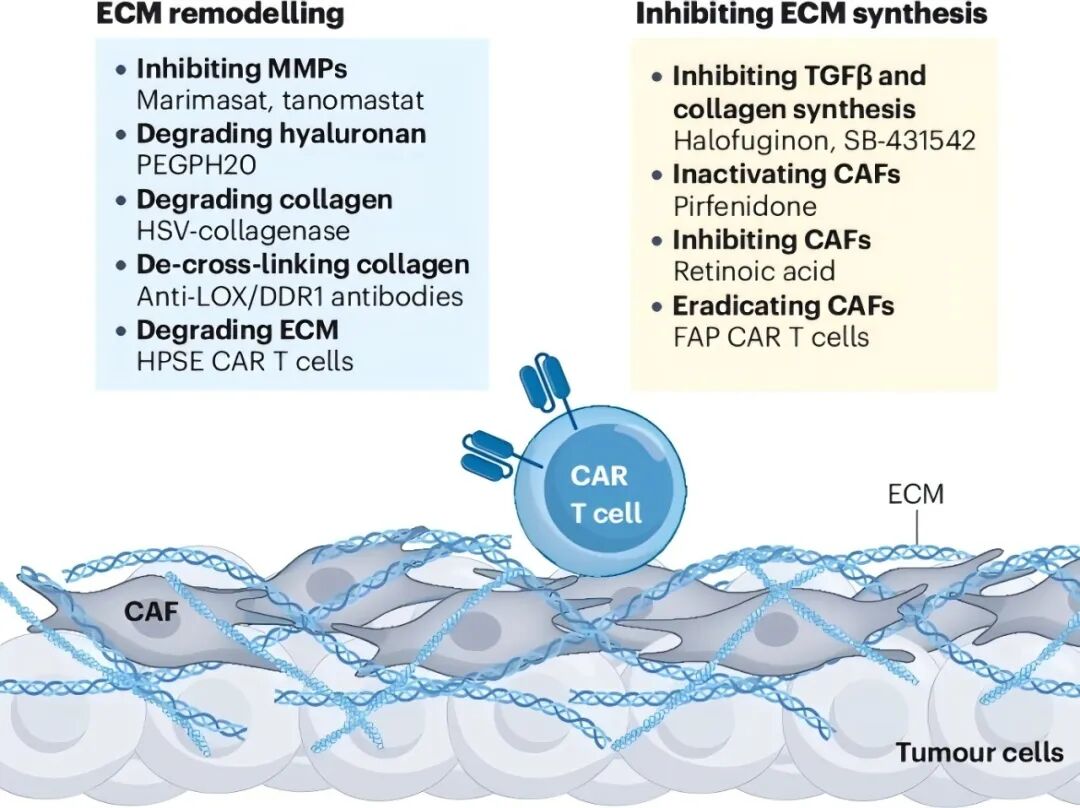
图 3:针对 ECM 的治疗策略
三、TME 中的抑制性免疫细胞
TME 内大量存在的抑制性免疫细胞(如 Tregs、MDSCs、抑制性 TAMs)构成了强大的免疫屏障。
这些抑制性细胞通过多重机制削弱 CAR-T:
其一是数量压制,稀释效应 T 细胞的比例;
其二是功能抑制,通过上调 PD-L1/CTLA-4 等免疫检查点分子以及分泌 IL-10、TGF-β 等抑制性细胞因子,直接诱导 CAR-T 细胞耗竭;
其三还涉及代谢竞争,加剧精氨酸耗竭、缺氧和酸中毒等恶劣条件,进一步损害 CAR-T 细胞的活力和功能。
破解这一免疫屏障需要采取综合措施:
一方面可阻断招募通路(如靶向 CCR4/CCR8)以减少抑制性细胞向 TME 的浸润;
另一方面可直接靶向清除 TME 内的抑制性细胞群,例如使用特异性抗体或小分子药物。
四、CAR-T 细胞「铠装」
为了赋予 CAR-T 细胞更强的能力以在恶劣的 TME 中生存并发挥功能,「细胞装甲」策略被广泛开发和应用:
在细胞因子工程方面,通过改造使 CAR-T 能够分泌 IL-18 或 IL-15,或引入显性负性 TGFβ 受体甚至利用 CRISPR 技术敲除 TGFBR2 基因,以阻断抑制性信号。
针对免疫检查点,策略包括采用显性阴性 PD1 受体以避免耗竭,或将 PD1 的胞外识别域与 CD28 的胞内激活域融合构建成「转换受体 」,巧妙地将抑制信号转化为激活信号。
面对代谢挑战,可通过过表达 GLUT1/GLUT5 等转运蛋白增强营养摄取能力,过表达 GPX4 以清除有害的脂质过氧化物,或联合使用 AMPK 激动剂与雷帕霉素等药物来提升细胞在缺氧环境下的耐受能力。
然而,装甲策略常伴随风险增加,且单策略的效果往往有限。因此未来的方向在于:开发时空精准调控技术、集成多重装甲、以及探索更有效的联合疗法。
五、调节 TME 线索以调整 CAR-T 细胞
肿瘤微环境(TME)的抑制特性可被逆向利用为 CAR-T 治疗优势,通过工程化改造,CAR-T 细胞能主动「劫持」TME 中的生物线索实现精准靶向与安全增效:
趋化因子受体重编程,使 CAR-T 细胞表达 CCR8、CXCR2 等原本富集于免疫抑制细胞的受体,显著提升实体瘤归巢能力;
缺氧响应系统,利用 HIF-1α 启动子或氧敏感结构域,将 CAR 活性严格限定于肿瘤低氧区域,规避对正常组织的脱靶毒性;
双信号门控设计(如 SynNotch 系统)则通过 ECM 成分等「信号 1」触发肿瘤抗原 「信号 2」的识别,将激活条件锁定于双信号共存的肿瘤微环境(图 4)。
这三类策略从本质上重构 CAR-T 与 TME 的互动关系 —— 将传统疗法中的障碍转化为定位导航、安全开关和智能激活器,在增强瘤内浸润深度的同时,将治疗窗口扩展至缺乏绝对肿瘤特异性抗原的难治癌种。
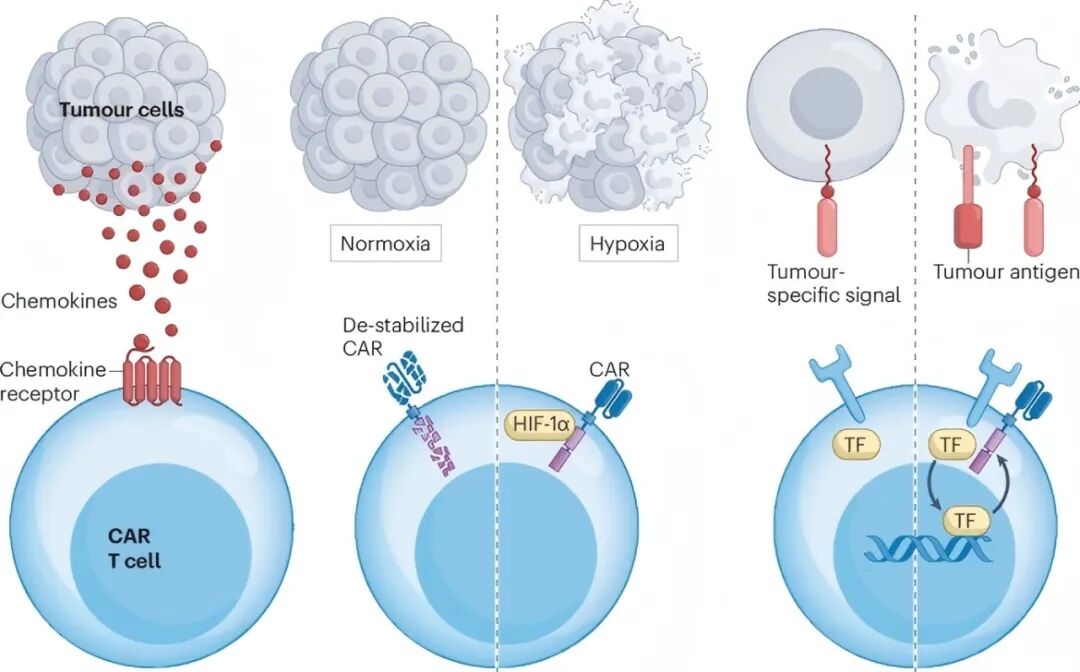
图 4:劫持 TME 线索以增强 CAR T 细胞的功效和安全性。
六、结论
实体瘤微环境(TME)的高度复杂性与动态演化本质,要求通过多靶点协同策略突破 CAR-T 耐药壁垒,而临床转化正面临毒性控制(如 CRS 风险)与 TME 异质性的双重挑战。
对此,Carl H. June 教授提出破局需践行「双向变革」:一方面重编程免疫抑制微环境(如逆转基质细胞表型),另一方面开发整合导航-感应-激活逻辑门控的智能 CAR-T。最终,通过「精准诊断 × 智能工程 × 联合疗法」三维范式的系统性整合,为实体瘤开辟治愈新路径。
来源:丁香通研选
我们长期为科研用户提供前沿资讯、实验方法、选品推荐等服务,并且组建了 70 多个不同领域的专业交流群,覆盖 CAR-T 工艺、肿瘤免疫、基因编辑、外泌体、类器官等领域,定期分享实验干货、文献解读等活动。


