


随着分子生物学技术的发展,检测基因结构和突变的方法不断涌现。尤其是 PCR 技术问世以后,各种与 PCR 相结合的基因检测技术进一步推动了基因研究的发展。如不对称 PCR 产物的直接测序 、核糖核酸酶酶切法 (Ribonuclease cleavage,RNAase)、限制性片段长度多态性分析 (Restriction Fragment Length Polymorphism,RFLP) 等等已成为基因分析的有力工具。但这些方法操作比较繁琐,局限性较大,或要求实验条件高,不适合一般临床实验室使用。1989 年问世的 PCR-SSCP(下文称 SSCP) 作为检测基因突变的方法,经不断地改进和完善,更为简便、快速、灵敏,不但用于检测基因点突变和短序列的缺失和插入,而且还被用于 DNA 定量分析,监测 PCR 诊断实验中的交叉污染情况,以及传染源的调查等。由于 SSCP 的突出的优点,近几年被大量地应用。一、SSCP 的原理及特点日本 Orita 等研究发现,单链 DNA 片段呈复杂的空间折叠构象,这种立体结构主要是由 其内部碱基配对等分子内相互作用力来维持的,当有一个碱基发生改变时,会或

Southern 杂交是分子生物学的经典实验方法。其基本原理是将待检测的 DNA 样品固定在固相载体上,与标记的核酸探针进行杂交,在与探针有同源序列的固相 DNA 的位置上显示出杂交信号。通过 Southern 杂交可以判断被检测的 DNA 样品中是否有与探针同源的片段以及该片段的长度。该项技术广泛被应用在遗传病检测、DNA 指纹分析和 PCR 产物判断等研究中。但由于该技术的操作比较烦琐、费时,所以现在有一些其他的方法可以代替 Southern 杂交。但该技术也有它的独特之处,是目前其他方法所不能替代的,如限制性酶切片段的多态性 (RFLP) 的检测等。一、基因组 DNA 的制备(前述)二、基因组 DNA 的限制酶切根据实验目的决定酶切 DNA 的量。一般 Southern 杂交每一个电泳通道需要 10-30 μg 的 DNA。购买的限制性内切酶都附有相对应的 10 倍浓度缓冲液,并可从该公司的产品目录上查到最佳消化温度。为保证消化完全,一般用 2-4 U 的酶消化 1 μg 的 DNA。消化的 DNA 浓度不宜太高,以 0.5 μg/μl 为好。由于内切酶是保存在 50% 甘油内的

第一个用于研究细胞骨架的药物,它是真菌分泌的生物碱。细胞松弛素(细胞松弛素 B 及其衍生物)在细胞内同微丝的正端结合, 并引起F-肌动蛋白解聚,阻断亚基的进一步聚合。当将细胞松弛素加入到活细胞后,肌动蛋白纤维骨架消失,使动物细胞的各种活动瘫痪, 包括细胞的移动、吞噬作用、胞质分裂等。它对微管没有作用, 也不抑制肌收缩, 因肌纤维中肌动蛋白丝是稳定的结构, 不发生组装及解聚的动态平衡。

流式细胞术(Flow Cytometry,FCM)是一种对液流中排成单列的细胞或其它生物微粒(如微球,细菌,小型模式生物等)逐个进行快速定量分析和分选的技术。 作为应用流式细胞术进行检测的技术平台,现代流式细胞仪产生于上世纪六七十年代。经过近四十年的发展和完善,今天的流式细胞仪已经十分成熟,并被广泛的运用于从基础研究到临床实践的各个方面,涵盖了细胞生物学、免疫学、血液学、肿瘤学、药理学、遗传学及临床检验等领域,在各学科中发挥着重要的作用。 流式细胞术-概述一种在液流系统中,快速测定单个细胞或细胞器的生物学性质,并把特定的细胞或细胞器从群体中加以分类收集的技术。其特点是通过快速测定库尔特电阻、荧光、光散射和光吸收来定量测定细胞 DNA 含量、细胞体积、蛋白质含量、酶活性、细胞膜受体和表面抗原等许多重要参数。 根据这些参数将不同性质的细胞分开,以获得供生物学和医学研究用的纯细胞群体。目前最高分选速度已达到每秒钟 3 万个细胞。 现代流式细胞术综合了流体力学技术、激光技术、电子物理技术、光电测量技术、计算机技术、荧光化学技术及单克隆抗体技术,是多学科多领域技术进步的结晶。随着现代科技的高速发

组成和普通显微镜一样,只不过物镜与照明系统颠倒,前者在载物台之下,后者在载物台之上,用于观察培养的活细胞,具有相差物镜。莱卡倒置显微镜进入 20 世纪 80 年代以来,光学显微镜的设计和制作又有了很大的发展,其发展趋势主要表现在,注重实用性和多功能方面的改进。在装配设计上趋于采用组合方式,集普通光镜加相差、荧光、暗视野、DIC、摄影装置于一体,从而操作灵活,使用方便。

细胞培养品名:细胞培养拼音:xi bao pei yang英文名称:culture of cells说明:包括微生物细胞、植物细胞和动物细胞培养。将动植物组织或细胞从机体取出,分散成单个细胞或直接以单细胞生物,使其在含有必要生长条件的培养基或培养瓶中继续生长与增殖。细胞培养是细胞生物学研究方面十分重要的技术。细胞周期及其调控、癌变机理、衰老、基因表达与调控等重要进展都离不开细胞培养技术。植物细胞培养为植物育种开辟了一条崭新的途径。大规模的细胞培养为营养品、疫苗生产以及药物研究、开发与肿瘤防治提供了全新的手段。原核生物、病毒培养为基因工程等现代生物工程提供了必要条件。细胞培养的基础知识细胞培养(cell culture)细胞培养的含义,简单地说即是把来自机体的组织经分散成为单个细胞,放在类似于体内的体外环境中生存,使其不断生长、繁殖或传代,借以观察细胞的生长、繁殖、衰老等生命现象。还可以利用细胞进行细胞工程与细胞癌变等重大问题的研究。细胞培养也是研究病毒与研制疫苗的基础技术。因此细胞培养技术在遗传学、免疫学、肿瘤学、病毒学、分子生物学等领域已得到广泛的应用。下面介绍原代培养和传代培养的基
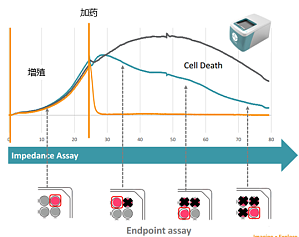
-基于阻抗方法实时、无标记、长期监测细胞表型- Maestro Z基于阻抗检测的细胞分析技术,使得实时、不间断且无需标记的细胞监测成为可能。对数据流的后续分析可以揭示细胞间互作和细胞-药物反应的动力学,可以更好地理解其机理而无需做费时费力的多次终点法实验。(可用于细胞增殖、肿瘤免疫、细胞毒性及活力检测、药物筛选、信号通路(GPCR/CFTR)、细胞间相互作用 (屏障功能)、病毒学研究及细胞迁移等细胞表型研究。) 细胞表型是涉及基因和蛋 ...

RT-PCR 和 qPCR 是大多数实验汪在科研道路上需要做的实验,大家看到这儿一定不以为意,这不就是两个简单的小实验,看两次 protocol,后面都不需要操作指南也能记住步骤的。可是…记住了实验步骤就真的等于能做好实验?能得到想要的数据、漂亮的曲线吗? 看似简单的实验,实则暗流汹涌,稍不注意的地方就可能有旋涡把实验前进的小船打翻,要不怎么会有那么多人为此悲催的实验结果整日愁眉苦脸,屡战屡败,屡败屡战……最后还是得战战战! 其实,多注意点小细节就可能帮助实验汪们减轻这样的苦恼,少做一些不必要的重复劳动,从而一战成功(即使不是一战,也少了 n 战)。 RT-PCR 虽然只是逆转 RNA,但却是决定后续 qPCR 或其他实验成功的关键因素,是重中之重。以下几点 tips 请查收: 1. 提取好的 RNA,先检测质量对 RNA 质量的检测包含 RNA 完整度测定及 RNA 产量测定两部分:(1)RNA 的完整度对 cDNA 的合成结果会产生重要的影响。通过琼脂糖凝胶电泳进行检测是方法之一,这也是一般实验室最常用的方法。通常完整的真核 RNA 应包括 28S、18S 及 5S RNA 条带

PCR(Polymerase Chain Reaction) 即聚合酶链式反应,对于常年狂奔在实验室的实验狗来说并不陌生。那么,对于 PCR 反应的核心成份,DNA 聚合酶,你选对了吗? 常见的 DNA 聚合酶如 Taq、Phanta、Pfu、KOD、Primerstar、Klenow、Bst、Phi29 等等。根据耐热性来分可分为耐热酶和常温酶。耐热聚合酶如 Taq、Pfu、KOD、Primerstar 等,常温聚合酶包括 Klenow、Bst、Phi29 等。根据保真度来分,高保真聚合酶 Pfu、KOD、Phanta 等,普通聚合酶 Taq 等。 面对如此之多的选择,大多数刚进实验室的新生们默默表示,选择恐惧症,犯了…… 那么,解决的方法是,根据实验目的选择合适的 DNA 聚合酶。比如:常规的 PCR 扩增和菌液鉴定,长度小于 5kb 的片段,保真度要求不高,使用 Taq 酶扩增即可。选用 Mix 的形式,混样更加简单,操作更加便捷。如果混样泡沫多,想灭掉泡沫的话,推荐大家使用 Green Taq Mix。想要获得较高的产量,可以选择 Taq Plus DNA 聚合酶,相对于 Ta

样本想要成功转化成为 PCR 的结果,前期需要注意的步骤有很多,包括:样本的采集、运送、保存以及核酸提取,每一个步骤都需要小心翼翼,中间的过程可谓是困难重重。样本在转化成 PCR 结果的过程中,究竟是哪些因素导致转化过程如此艰难呢?下面由小编带着大家一起去扒一扒样本中影响 PCR 检测的抑制物。 图 1. 核酸处理与检测流程图 一、PCR 抑制物的来源PCR 抑制物的来源包括内源性与外源性两种1. 内源性天然存在标本中的组成成分。如:免疫球蛋白、蛋白酶、血红蛋白及其代谢产物、血红蛋白中的乳铁蛋白、肌红蛋白、脂类、黏蛋白、尿素、离子、胆盐、多糖等。 2. 外源性外部引入的抑制物。如:肝素抗凝剂、纤维素和硝酸纤维素、手套滑石粉、标本容器或采样器材上含有的抑制物 二、PCR 抑制物一般作用机制PCR 抑制物中大部分的作用机制尚不完全清楚,但基本可以归为三类:1. 干扰核酸提取过程中的细胞裂解;2. 降解或包裹核酸;3. 热稳定 DNA 聚合酶失活。 1. 干扰核酸提取过程中的细胞裂解 细胞裂解,核酸释放使得核酸与酶作用产生扩增。如果细胞裂解不完全,核酸释放不完全,就不会产生扩增。常见的简单处
荧光定量 PCR 的常见异常结果除了扩增曲线异常、融解曲线异常两类常见情况外,qPCR 实验中通常还会遇到以下三大类情况: 1. 复孔间重复性差怎么办?复孔间重复性差一般会有以下两种情况:(1)CT 值很大,如 CT≥ 30,重复性差属于正常现象。该现象符合泊松分布,即在有效模板量很少的情况下,模板与引物的碰撞存在随机性,直接导致复孔间的 CT 值差异较大。解决方法:如果融解曲线没有杂峰,无模板阴性对照同目的基因的 △CT 值为 3 or 5 以上,那 CT 值为准确的,可多设置几个复孔,选择重复性好的 CT 值参与计算。(2)CT<30,重复性较差。这种情况一般同操作有关。解决办法:从以下几个方面进行问题的排查:①加样准确度;②移液器吸取液体的准确度;③定期校准 qPCR 仪。 ①加样准确度避免小体积加样,减少加样误差。可以将引物、SYBR Green Mix、ddH2O 配置成混合体系,并且增加模板的稀释梯度,大体积加样。如:原液 cDNA 添加 1μl,可以更改为原液 cDNA 稀释 5 倍,添加 5μl,使用 ddH2O 补齐体系至 20μl 即可。SYBR Green Mi
实时荧光 PCR(real-time PCR)因其全封闭扩增,简单快速,重复性好,无扩增后处理以及易于自动化等优点,广受大家欢迎。在分子诊断领域中,实时荧光 PCR 方法涉及的领域包括感染性疾病、肿瘤、遗传病等多方面。那么实时荧光 PCR 技术是如诞生的呢?下面,由小编带领大家一起了解大牛们是如何经过精密的研究一步一步探索出来这项技术。1983 年1983 年美国科学家 Kary Mullis 发明 PCR 方法,用于放大扩增特定的 DNA 片段,可看作是生物体外的特殊 DNA 复制。1985 年1985 年,Cetus 公司从温泉中分离的嗜热菌(Thermus aquaticus)株中纯化出耐热 DNA 聚合酶(后面简称 Taq 酶)。该酶耐高温的性质极大地提高了 PCR 扩增的效率,使得 PCR 真正变为现实,为其自动化铺平了道路。但是 PCR 技术也有其相应的局限性,就是对扩增产物分析时需要开盖处理,容易造成污染。为了解决这一问题 Russ Higuchi 进行了一系列相关研究。1992 年1992 年,Higuchi 在一次报告中提出了实时荧光定量 PCR 技术。通过检测溴化乙

PCR 过程中免不了遇到各种问题,如扩不出目的条带、有非特异性条带、空白对照出现扩增产物、扩增产物跑胶弥散或拖尾、高保真 PCR 出现突变等等。今天师兄就带大家来聊一聊 PCR 的常见问题应该如何解决。 PCR 实验包括反应体系的配置和反应程序,反应体系中有模板、引物、dNTP、酶、buffer、Mg2+,反应程序又包括预变性、变性、退火、延伸和彻底延伸。那么在 PCR 出现问题时主要是从反应体系的各个组分和反应程序上来进行调节优化。 Q1: 实验组无扩增条带A1:(1) 引物检查人工合成的引物是否因存储条件不当而降解(建议用新合成的引物进行尝试);引物设计是否合理,可利用BLAST检查引物特异性或重新设计引物(如果之前用过该引物,可排除引物设计问题)。 (2) 模板长期放置、反复冻融会导致模板断裂、开环或降解,应使用新鲜制备的 DNA 双链作为模板;模板 GC 含量过高会导致 DNA 的双链无法打开,此时加入 PCR Enhancer,可以有效降低解链温度;模板为粗品,有可能是 DNA 未释放出来(若样本为植物叶片,要确保植物为非多糖多酚植物,取新鲜幼嫩的叶片,并将叶片面积
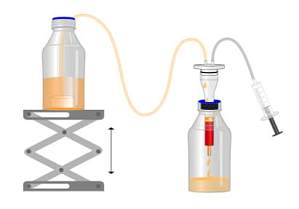
在哺乳动物和昆虫细胞表达系统中,目的蛋白经常分泌到细胞培养基。因此,在进行纯化前,我们的目的蛋白已存在于大体积培养液中。然而有时,用来纯化目的蛋白柱子体积却只有 1 ml。时间金贵,手动一次一次的将样品加入柱子中就太麻烦了,简直是要加到头秃有木有! 不知道大家为了简化操作,节省时间,都使用了怎样的方法来上样呢? 今天为在这里为大家分享一个锦囊工具:Wet Fred。这个小体积的工具非常灵活,可以根据我们的实验需要,使用在实验台、冰箱和冷室中。 优点Wet Fred 工作原理是静水压力(虹吸原理),适配 IBA 的 Strep-Tactin® /XT 重力柱。将培养液瓶和液体收集瓶通过这个工具连接起来,拉动推塞,通过调节高度来调节流速,就能达到「自动」上样啦。 1. 操作简单2. 可以过夜,也可以放在冰箱中3. 不会堵塞4. 柱子不会变干燥5. 适合体积大的样品 这种不用在旁监督的上样小工具,简直是解放双手的利器!
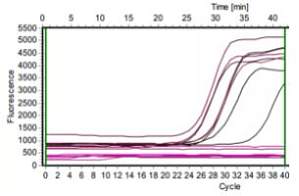
还在为奇怪的结果发愁吗?老板问起来我该怎么答?我该如何搞定我的曲线?想解决 qPCR 的曲线难题,请听小 V 细细讲解~qPCR 曲线异常问题通常分为两大类:扩增曲线异常、融解曲线异常。 扩增曲线异常这个问题可能涉及到多种原因,主要可以归结为以下 6 种:(1)扩增曲线不光滑主要有两个原因,一是扩增信号太弱,经系统矫正后,就产生这样抖啊抖的线,小 V 建议大家提高模板浓度重复实验。二是仪器本身的问题,可能在扩增过程中仪器出现了波动或者本身该检测孔出现了异常。 (2)扩增曲线断裂或下滑比较典型的曲线下滑如上图所示,主要原因是模板浓度太高了,在基线期内就起峰了,仪器会默认起峰的线条仍为基线部分,将扩增曲线往基线位置下拉,因此你的曲线就趴下来啦~这种情况大家也不要慌,可以减小基线终点(例如基线期默认 3 - 15 个循环,改为 3 - 10 个循环),重新分析数据,一般都能得到一个比较好的结果。 (3)个别扩增曲线骤降这是比较常见的一个问题,反应管内若留有气泡,温度升高后会导致气泡破裂,使仪器检测到的荧光值突然降低。所以大家进行扩增反应之前,一定要仔细检查反应管内是否有气泡残留。 (4)扩增
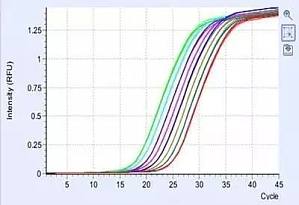
充分的准备是实验成功的关键,那么,qPCR 实验需要提前做好哪些了解呢? 试剂的储存条件市面上大多数 qPCR 试剂推荐 - 20 ℃ 避光长期储存。Vazyme 的 ChamQTM 和 AceQ® 两个系列的 qPCR Master Mix,长期储存应置于 - 20 ℃ 避光,解冻后可于 4 ℃ 短暂存放;避免反复冻融。 原始模版稀释倍数尽量选择 Ct 值落在 15 - 28 范围的稀释倍数作为原始模板稀释度。 反应体系与次数推荐 20 μl 体系,Vazyme ChamQTM/AceQ® QPCR Master Mix 5 ml 可供 20 μl 体系 500 次反应。 模板的上样量若直接使用 cDNA 原液作为模板,建议使用体积不超过 qPCR 反应总体积的 1 / 10!当复孔之间重复性不佳时,可将模板做适当稀释后以大体积加入反应体系,可有效提高实验重复性。看小 V 做的实验,是不是重复性爆表啊~~~ 预变性时间常见的 qPCR 试剂预变性时间从 30 sec - 15 min 不等。科学家们为了封闭常温下酶的活性,使 qPCR 产生非特异性扩增的概率降到最低,想出了这种只
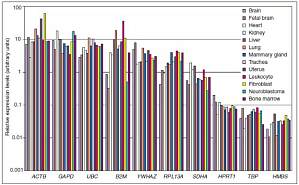
荧光定量 PCR 的主要用途之一是相对定量,而提到相对定量,必然离不开内参,那什么是内参基因?为什么相对定量必须使用内参?如何选择正确的内参基因?今天,我们就来聊聊关于内参的那些事。 什么是内参?内参基因,也称为管家基因,其编码蛋白是维持细胞基本生命活动所必须的蛋白质。它的表达水平不受任何内源性与外源性因素的影响。管家基因高度保守且在大多数情况下持续表达,稳定表达于不同类型的细胞和组织中。 为什么相对定量必须使用内参?那内参基因与相对定量又有什么样的关系呢?我们通过以下实验案例一起了解一下。实验目的:测定肝癌细胞 X 基因相对于正常肝细胞的表达量。通过荧光定量检测可得:肝癌细胞中 X 基因的 CT= 25,正常肝细胞中 X 基因的 CT= 26,所以,利用表达量倍数计算公式 2-△CT计算,发现肝癌细胞中的 X 基因表达量是正常肝细胞表达量的 2 倍。但是,上面这个定量结果检测有效的前提是:必须保证两盘细胞的细胞数量完全一致;RNA 提取、逆转录以及定量效率及操作必须完全一致;不存在操作误差等。这显然是无法达到的。 那如何才能使这些误差不影响 X 基因表达检测的真实性呢?这时,为了将样
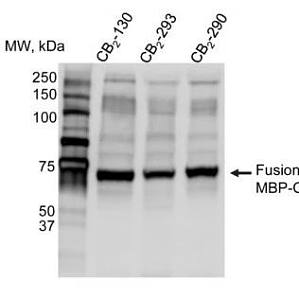
膜蛋白在生物体的许多生命活动中起着非常重要的作用,如细胞的增殖和分化、能量转换、信号转导及物质运输等。据估计大约有 60% 的药物作用靶点是膜蛋白。大多数膜蛋白的含量较低, 另外用于溶解细胞膜上 GPCR 的高浓度洗涤剂进一步限制了下游的纯化方法。传统的色谱方法,如离子交换或疏水色谱,对于从稀释的含清洁剂的溶液中纯化大量蛋白质是无效的。膜蛋白的纯化仍是目前很多科研工作者遇到的难题之一。今天,我们就跟大家分享一篇膜蛋白纯化实例。人大麻素受体 CB2 是一种完整的膜 G 蛋白偶联受体 (GPCR),主要表达于免疫源细胞。它涉及炎症、神经退行性疾病、肥胖和疼痛等代谢途径的调节,因此也是药物开发的一个重要靶标。在本篇文章中,作者使用亲和标签开发的新纯化方案能够有效分离宿主细胞中低表达的重组 GPCR。该方法适用于制备毫克量的稳定同位素标记受体,用于高分辨率核磁共振研究。 一、实验设计思路1、CB2 表达载体构建:为了找到最合适的标签位置,作者对 Linker 和 Twin-Strep-tag 标签位置进行了不同的设计。2、E.coli 系统表达 CB2 蛋白及膜蛋白制备,测定 strep-ta
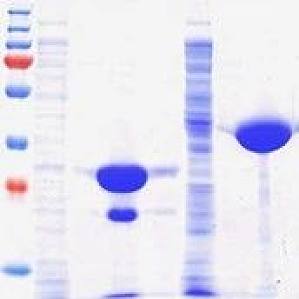
无论是出于科研还是工业目的,成功的表达并且纯化所需的蛋白,都是进一步进行分析和研究的基础。在质上和量上有所不同,选择一个合适的纯化系统,来生产并纯化所需质和量的蛋白,值得慎重考虑。一套合适的纯化系统,不仅可以节省时间和成本,更可以节省蛋白样品。 亲和层析法将目的蛋白从其他细胞组分中纯化出来的方法多种多样,目前,最常用的方法之一是亲和层析法。在其中,通常的方法是将目的蛋白与一段特殊的短肽或蛋白,也就是亲和标签所融合。这种亲和标签针对配体有特殊的亲和力。而配体可以是固定在层析基质上的其他蛋白质,小分子或是金属。当融合有亲和标签的蛋白通过标签-配体作用结合再层析基质上时,通过洗杂步骤,即可去除掉其他的细胞成分。为了洗脱所需要的蛋白质,通过改变缓冲液的条件,如 pH,或通过竞争亲和标签和配体连接的方法来进行。 但怎样的纯化是较为成功的呢?仅达到所需的质量量级,如毫克级(或克级)即可满足吗?这些并不简单。质在蛋白纯化中同样尤为重要,比如,就蛋白的生物活性和纯度而言,通常会有所要求。以下我们来讨论并且比较 2 种技术:His-tag 系统和 Strep-tag® 系统,都使用了亲和层析法来纯化得到
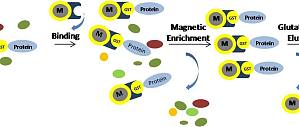
蛋白纯化的方法很多,如层析法、电泳法、超离心法、超滤等,其中蛋白质亲和层析法通常只需要一步操作便能将目标蛋白从混合物中分离出来,且纯度很高,因而备受实验者的青睐。在进行蛋白表达时,选择合适的标签有利于蛋白的纯化,促进蛋白的可溶性,因此了解几种常用的蛋白纯化标签很重要。一般来说,常用的蛋白纯化标签主要有 His tag、GST tag、MBP tag、NusA tag、Strep tag 等,那么这些蛋白纯化标签有什么不同之处呢?His tag(组氨酸标签) 融合蛋白是目前最常用的表达方式,其优点是标签小,纯化步骤简便,纯化条件温和,能纯化可溶性/包涵体蛋白,一般不会影响蛋白的功能结构,且可以纯化出大量的目标蛋白,但该标签不适合易氧化蛋白或膜蛋白的纯化。GST tag(谷胱甘肽巯基转移酶) 的洗脱条件温和,有助于保持蛋白功能活性,适合 pull-down 检测,具有很好的线性动态范围。但缺点是分子量较大,可能会影响蛋白质的功能和下游实验。另外,如果蛋白不可溶,很难用变性的方法进行纯化。 MBP tag(麦芽糖结合蛋白标签) 可以减少目标蛋白的降解,增加蛋白的表达量和稳定性,提高表达产物的
关于丁香通
公司信息
个人用户
企业机构


