Quantitatively Profiling Genome-Wide Patterns of Histone Modifications in Arabidopsis thaliana Using ChIP-seq
互联网
465
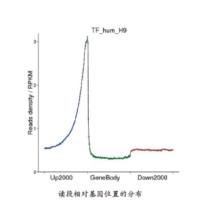
Genome-wide quantitative profiling of chromatin modifications is a critical experimental approach to study epigenetic and transcriptional control mechanisms. Since first being reported in 2007, chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) has soon became a popular method-of-choice for profiling chromatin modifications and transcription factor-binding sites in eukaryote genomes. ChIP-seq has the advantage over the earlier ChIP-chip approach in multiple aspects including the lower amount of input DNA required, an expanded dynamic range and compatibility with sample multiplexing. Here we describe a detailed protocol for profiling histone modification in the Arabidopsis thaliana genome with ChIP-seq using the SOLiD™ 2.0 high-throughput sequencing platform. As read length and sequencing depth are two critical factors determining data quality and cost, we have developed bioinformatics approach to evaluate the effect of read length and sequencing depth on the alignment accuracy and the generated chromatin profile, respectively. Our analyses suggest that 2–3 million high quality sequencing tags with a read length of 35 nucleotides would be sufficient to profile the majority of histone modifications in this popular model plant species.