

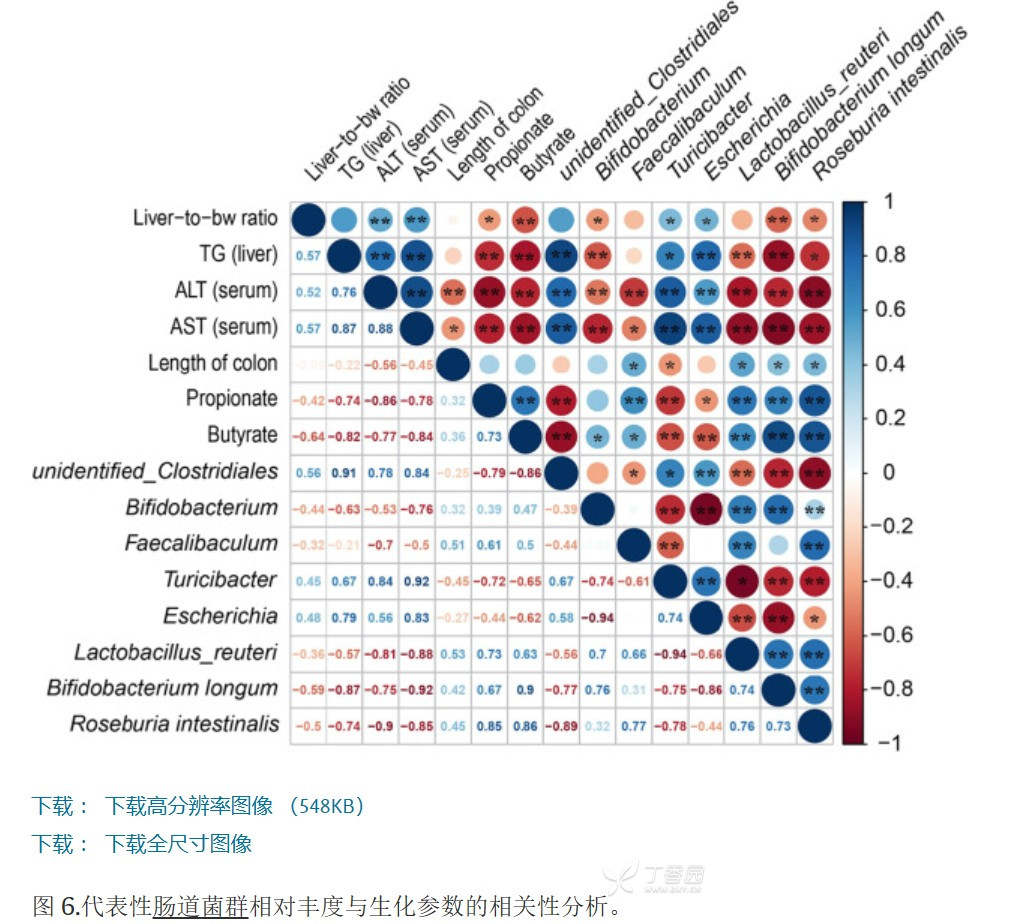
 请问大家,这种肠道菌群与机体生化指标的相关性的图,请问是做16s之后就可以自己分析出来的,还是得做宏基因组得到呢?
请问大家,这种肠道菌群与机体生化指标的相关性的图,请问是做16s之后就可以自己分析出来的,还是得做宏基因组得到呢?3 个回答
huarenqiang5
有帮助
这个一般还是需要做宏基因组而得到。
loveliufudan
有帮助
斯皮尔曼分析的数据可以从16S测序数据中获得。16S测序是一种常用的方法,用于分析微生物群落的组成和相对丰度。通过测序肠道样品中的细菌16S rRNA基因片段,可以获得关于不同细菌菌属相对丰度的信息。
在斯皮尔曼相关性分析中,你可以将菌群的相对丰度作为一个变量,机体生化指标的数值作为另一个变量,然后计算它们之间的相关性。
宏基因组测序(metagenomics)是一种更全面的方法,可以提供关于微生物群落的基因功能和物种多样性的信息。如果你对微生物群落的功能和物种多样性与机体生化指标之间的关系感兴趣,那么宏基因组测序可能是更适合的选择。
综上所述,斯皮尔曼相关性分析可以使用16S测序数据中的菌群相对丰度和机体生化指标的数值进行。如果你对微生物群落的功能和物种多样性也感兴趣,那么宏基因组测序可能是一个更全面的选择。选择合适的分析方法应根据你的研究目标和资源可行性进行评估和决策。
土井挞克树
有帮助
得做宏基因组能得到,单纯分析16s得不到全部数据
相关产品推荐




相关问答
关于丁香通
公司信息
个人用户
企业机构

提问
扫一扫

实验小助手

扫码领资料
反馈
TOP
打开小程序