

- ¥2000
- 2025年07月16日





万千商家帮你免费找货
0 人在求购买到急需产品
- 详细信息
- 询价记录
- 文献和实验
- 技术资料
- 服务名称:
蛋白结构分析蛋白结构预测与药物分子设计服务
- 规格:
C3506-10G
提供服务:
1. 结构建模(蛋白突变亚型的结构建模)
2. 利用蛋白结构进行特定分子(配体,底物,结合蛋白)的停泊( dock)
3. 特定结构域上的小分子化合物配体的搜索。
反馈内容:
1. 结构:
根据pdb数据库确定是否有已知的结构,是否有同源蛋白或同源结构域的结构信息。
有已知蛋白三维结构,可以直接利用,或分析后进行利用
有同源序列,通过同源建模构造蛋白结构--模拟结构(即同源建模)
2. 结构分析
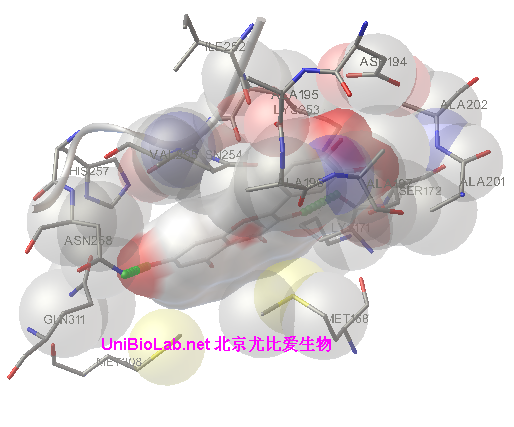
同源蛋白和突变蛋白的结构分析,突变氨基酸的位置,结构变化,势能变化,蛋白表面改变(结构,电荷性质等)。
3. 对蛋白设计亲和分子进行药物设计的候选物质结构设计.
4. 其他要求
团队介绍:
1. 团队由软件工程师和生物学专家组成. 利用权威性的一些方法和软件之外,还拥有自主开发的蛋白结构分析和编辑软件,
可以对结构进行自定义分析,可以适应一些新的思路和兴趣需要;
2. 指导突变体改造方案,对已知或未知配体进行安装和分析,确定小分子结合的部位,相互作用的氨基酸;根据结构设计蛋白的结合小分子等。
联系电话: 134-6651-0334 email:583348587@qq.com
北京尤比爱生物科技中心 www.unibiolab.com
风险提示:丁香通仅作为第三方平台,为商家信息发布提供平台空间。用户咨询产品时请注意保护个人信息及财产安全,合理判断,谨慎选购商品,商家和用户对交易行为负责。对于医疗器械类产品,请先查证核实企业经营资质和医疗器械产品注册证情况。
- 作者
- 内容
- 询问日期
 文献和实验
文献和实验AI 筛选 + 虚拟筛选:新药研发的 “双引擎” 的核心逻辑
智能 (Artificial Intelligence,AI) 药物筛选是一种结合 AI 技术与计算化学的高通量筛选方法,广泛应用于蛋白结构预测、新药研发和分子设计与优化等领域。其主要目的是利用机器学习 (MachineLearning,ML) 算法分析大量数据,从中学习规律,生成 AI 打分函数,以此提高筛选效率,加速候选药物的发现过程。 分子对接 www.medchemexpress.cn/molecular-docking-service.html 分子对接 (Molecular Docking)是一种基于计算模拟
大的 exhaustiveness。如果我们大致知道 binding pocket 在什么位置,那准确性应该会高不少,如何大致确定 binding pocket 的位置呢?我们接着试验。可以通过实验的方式,比如某个点突变对结合或者活性影响非常大,那么大概率这个残基是结合口袋的一部分。可以通过软件预测,比如蛋白与配体(底物)结合位点预测:https://zhanglab.ccmb.med.umich.edu/COACH/再比如 Discovery studio 软件(专业版的),可以很方便的根据受体分子的表面形状来预测结合
HNADOCK - 一个研究 RNA 与 DNA/RNA 互作的平台
提供要对接的两个分子的三维(3D)结构,其中 RNA 接受序列和 PDB 结构文件(或 PDB 登录号如 1KD5:A)的输入,而 DNA 仅支持 PDB 结构文件(或 PDB 登录号如 1KD5:A)的输入。用户还可以选择提供结合位点信息(HNADOCK 可以自动整合结合位点信息),以及选择是否优化前十个模型。指定结合位点残基可以使预测的模型准确性更高。用户可以提供两种类型的绑定站点信息。(i)结合位点残留在受体或配体上:在文本框中如下指定结合位点残基15:A,23 - 26:A,18:B代表
 技术资料
技术资料暂无技术资料 索取技术资料

