


- ¥600 - 2800
- 蛋白质定量DDA:适合差异分析吗?
- 全国
- 2026年07月19日

企业认证
相关产品推荐更多 >





万千商家帮你免费找货
0 人在求购买到急需产品
- 详细信息
- 文献和实验
- 技术资料
- 提供商:
北京百泰派克生物科技有限公司
- 服务名称:
蛋白质定量DDA:适合差异分析吗?
- 规格:
询价
蛋白质定量DDA可用于差异分析,其适用性主要取决于样本复杂度、实验条件一致性和研究目标。DDA采集会优先对信号较强的前体离子进行碎裂分析,同一肽段在不同进样中未必都能被稳定采集。对于蛋白组成复杂、丰度跨度较大的样本,低丰度肽段更容易漏检,缺失值也可能增加,进而影响组间比较。当各组样本来源一致、前处理流程统一,且研究目标以候选差异蛋白筛选为主时,Label-free DDA可兼顾蛋白鉴定和相对定量。若项目更关注低丰度蛋白的稳定检出、跨样本数据完整度或复杂分组比较,则需进一步评估DIA或TMT标记定量方案。
一、蛋白质定量DDA如何支持差异分析
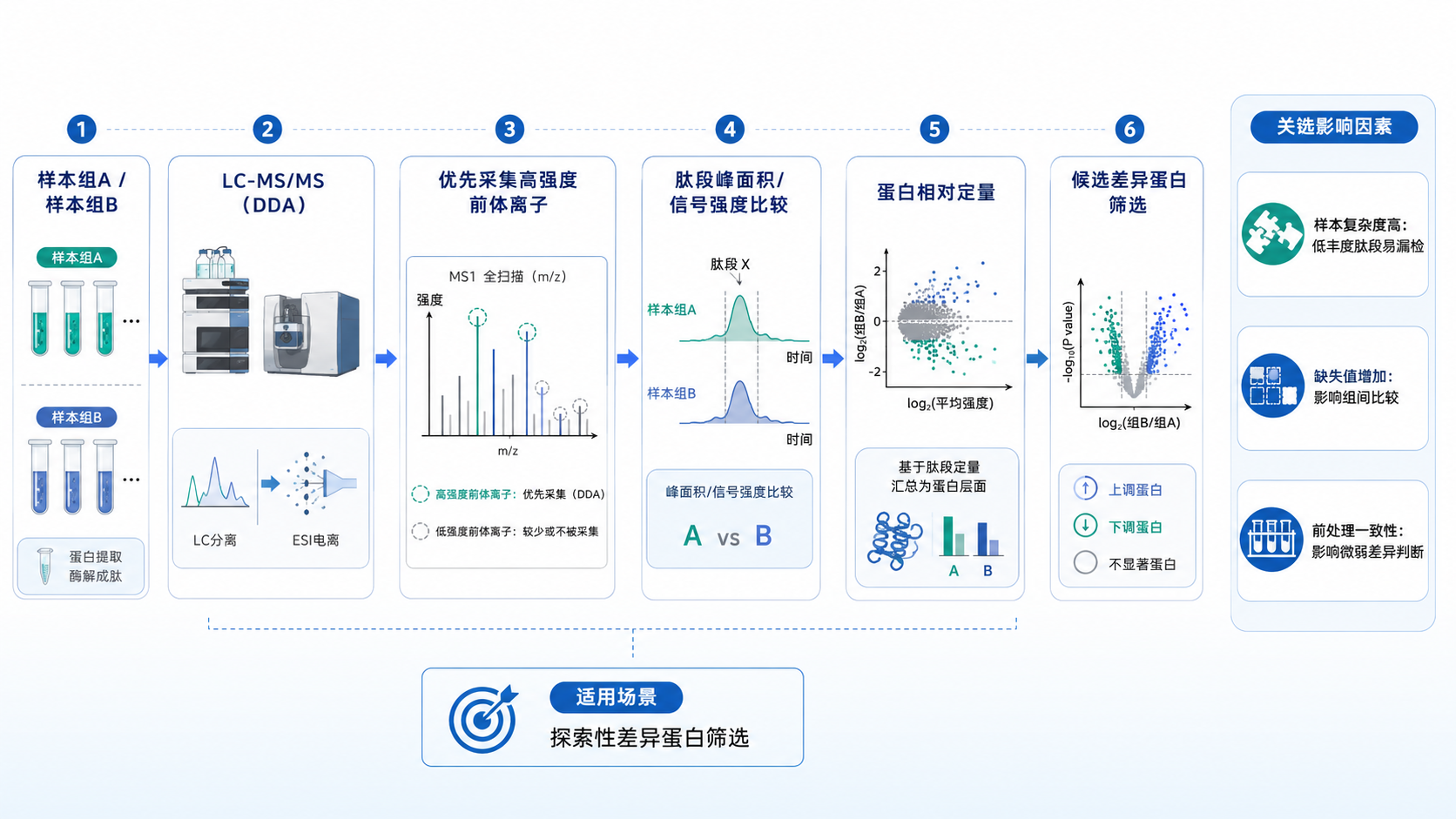
图1
1、定量结果来自肽段信号比较
(1) 比较前体离子信号
Label-free DDA通常比较不同样本中肽段前体离子的色谱峰面积或信号强度,再将多个肽段的定量信息汇总至蛋白层面,用于分析蛋白相对丰度变化。
(2) 肽段鉴定决定定量范围
肽段需要获得可靠鉴定,并在不同样本中完成信号匹配,才能进入蛋白定量。某一肽段未在部分样本中检出,并不等同于对应蛋白未表达,也可能与离子信号较弱、共洗脱或前体选择差异有关。
2、DDA适合探索性差异筛选
(1) 同时获得鉴定与定量信息
DDA可在蛋白鉴定的基础上获得相对丰度信息,适合分析样本中的蛋白组成、筛选候选差异蛋白,并为后续靶向验证确定范围。
(2) 样本设计较为灵活
Label-free DDA不受标记通道数量限制,适合样本数量可能调整或分组关系相对简单的探索性蛋白质组学项目。
二、DDA差异分析受到哪些因素影响
1、样本复杂度影响肽段检出
(1) 高丰度离子更容易被采集
组织、体液和细胞裂解物中的蛋白丰度差异较大。高丰度肽段产生的离子信号较强,更容易进入二级质谱,低丰度蛋白则可能缺少稳定的肽段证据。
(2) 缺失值影响组间比较
同一肽段若只在部分样本中获得定量,其变化可能同时受到生物学差异和采集波动影响。数据分析时需要结合缺失值分布、肽段检出情况和组内重复性进行判断。
2、实验一致性影响微弱差异判断
(1) 前处理与检测条件需要统一
样本裂解、蛋白提取、酶切、脱盐和进样过程中的差异,都可能改变肽段信号。项目设计时应根据具体样本体系统一前处理流程,并合理安排生物学重复和检测批次。
(2) 蛋白结果需要结合肽段证据
若某个蛋白仅由少量肽段支持,或不同样本中的定量结果来自不同肽段,不宜只依据差异倍数进行筛选。还需复核肽段检出一致性、色谱峰形、鉴定质量和组内变化趋势。
三、如何选择适合的蛋白定量方案
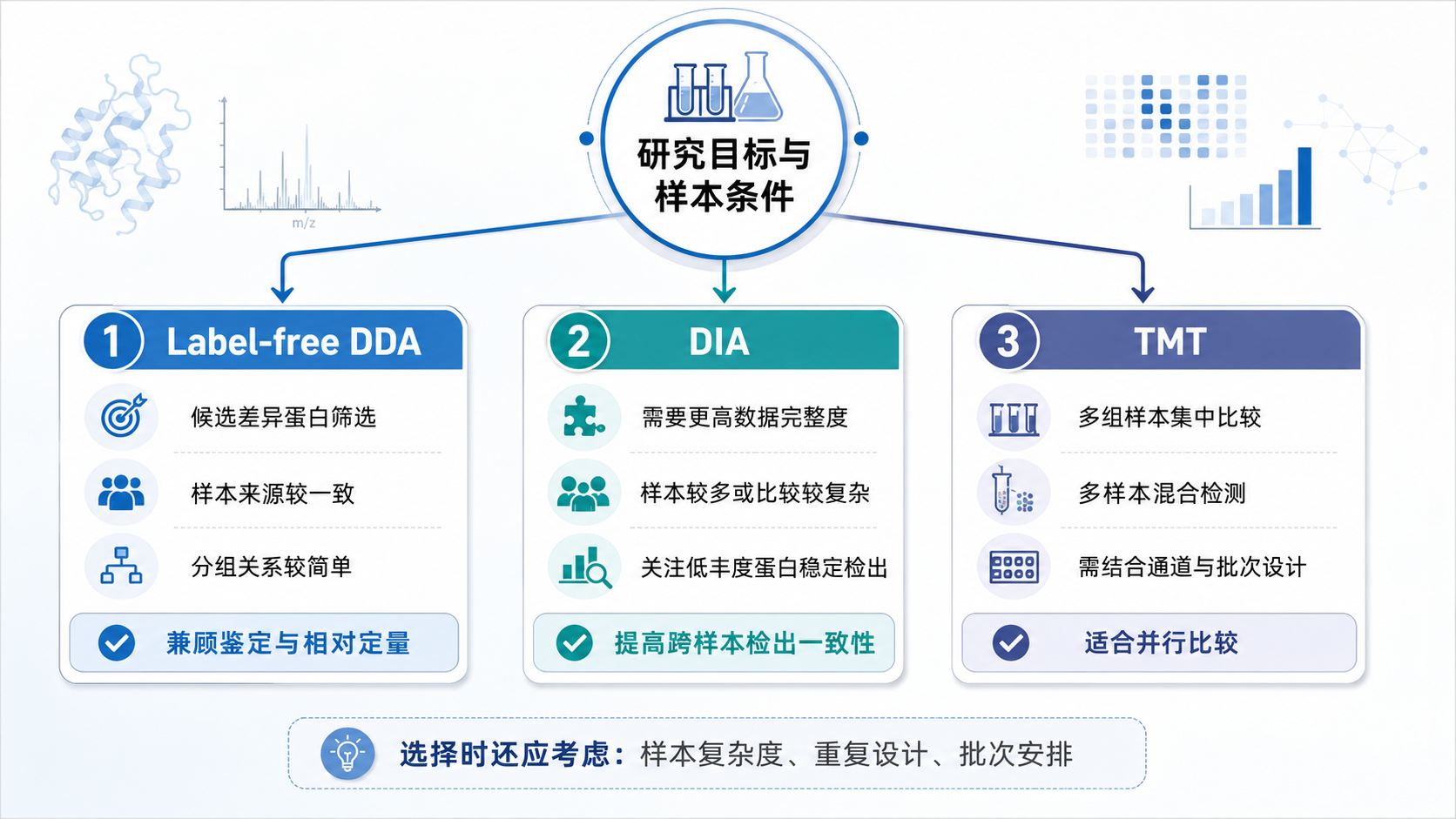
图2
1、适合采用Label-free DDA的项目
(1) 以候选差异蛋白筛选为主要目标
当研究目标是了解整体蛋白组成、寻找可能发生变化的蛋白并建立后续验证清单时,Label-free DDA可以满足探索性鉴定和相对定量需求。
(2) 样本条件和分组关系较为一致
若各组样本来源一致,前处理流程能够统一,且研究结论不依赖低丰度蛋白在所有样本中的连续检出,Label-free DDA具有较好的适用性。
2、需要评估DIA或TMT的项目
(1) 对跨样本数据完整度要求较高
DIA采用连续窗口采集一定范围内的离子信号,更适合样本数量较多、比较关系较复杂,或需要提高肽段跨样本检出一致性的项目。
(2) 需要开展多组样本集中比较
TMT通过同位素标签对多个样本进行标记和混合检测,适合多组样本的集中比较。方案设计时需结合样本数量、通道安排、批次关系及共分离干扰等因素进行评估。
蛋白质定量DDA适合用于探索性差异蛋白筛选,但结果解释不能只看差异倍数和统计值。缺失值分布、组内一致性、肽段证据和蛋白鉴定质量,共同决定候选结果是否具有后续验证价值。检测前明确样本条件、分组关系和研究重点,有助于选择与研究需求相匹配的定量路线。百泰派克生物科技可提供DDA蛋白质鉴定、Label-free定量蛋白质组学、DIA定量蛋白质组学、TMT定量蛋白质组学及差异蛋白分析服务,涵盖样品前处理、LC-MS/MS检测、生物信息学分析和结果报告等环节。如需开展DDA蛋白质鉴定或相关蛋白质组学研究,欢迎联系我们进行项目评估,检测路线与数据分析方案将结合样本条件、分组设计和研究目标确定。
风险提示:丁香通仅作为第三方平台,为商家信息发布提供平台空间。用户咨询产品时请注意保护个人信息及财产安全,合理判断,谨慎选购商品,商家和用户对交易行为负责。对于医疗器械类产品,请先查证核实企业经营资质和医疗器械产品注册证情况。
 文献和实验
文献和实验随着不同的基于质谱分析方法的发展,可以研究的蛋白互作网络的范围也在逐渐增加。更重要的是,用于肽和蛋白质定量的新兴方法产生的数据是完整且无偏差的,是研究完整网络或蛋白互作的基本要求。 基于数据依赖性采集(Data-dependent acquisition, DDA)易于执行,并能快速评估样本中的物质的数量。除了简单之外,还需要对蛋白质进行鉴定和翻译后修饰。任何蛋白质的生物学特性在动态系统中的已知程度都是极微小的,不断增加的拼接变异体,SNP等很可能意味着在可预见的未来,使用数据依赖性采集
超微量外泌体蛋白质组突破用量极限和检测上限-低至200μL血浆!高达4000+ EV蛋白!
变性以及MS/MS谱图的质量(信息含量)的可变性。最终结果是,来自不同生物样本的数据集没有识别和量化同一组蛋白质;或者换句话说,DDA模式的lable-free定量蛋白质组学数据集将包含很多缺失值,并且这主要影响较低丰度的蛋白质。近年来,数据非依赖性采集(data-independent acquisition,DIA)技术出现,指定m/z范围内的所有前体离子都同时进行MS碎片化,复杂的MS2谱图与包含碎片离子谱图、前体离子精确质量及其归一化保留时间的谱图库搜库比对。DIA在增加已鉴定肽的数量、提高定量精度和增加
-MS/MS系统使一次实验鉴定上万个蛋白成为可能。基于LC-MS/MS系统方案也叫shot-gun法,质谱的采集模式为DDA(Data Dependent Acquisition)。在这种模式下,质谱根据离子化的肽段母离子的强度,依次选择10到20个强度最大的离子进入串联质谱,进行进一步碎裂,然后经过与模板蛋白质数据库比较确认。这种方法无疑丢失了大量有用的肽段母离子信息。此外,经过二级碎裂的二级谱图,在后期使用软件鉴定中也仅有少量谱图得到解析(约30%),大部分谱图依然得不到有效利用。由于质谱选择离子的随机
 技术资料
技术资料暂无技术资料 索取技术资料

