- 移动端


粒曼生物科技(武汉)有限公司
3 年
手机商铺
- NaN
- 0
- 0
- 0
- 0
推荐产品




技术资料/正文
问题解读 I 为什么qPCR不完全适用于KO细胞的敲除效率评价?
266 人阅读发布时间:2024-09-12 19:21
1. qRT-PCR(qPCR)技术原理
RT–PCR(Reverse Transcription-Polymerase Chain Reaction),即逆转录-聚合酶链式反应,是以细胞中的mRNA作为模板,设计特异性引物将mRNA反转录成cDNA,再以cDNA为模板进行PCR扩增,从而获得目的基因的表达情况。qRT-PCR(Quantitative Reverse Transcription PCR,qPCR),即实时荧光定量PCR,是使用荧光探针(例如Taqman探针),在PCR过程中利用荧光信号的变化实时检测PCR扩增反应中每一个循环扩增产物量的变化,最终对起始模板进行精确的定量分析。
Taqman探针是由5’端标记有荧光报告基团和3’端标记有淬灭基团以及与目标基因特异性结合的寡核苷酸序列组成。探针完整时,报告基团发射的荧光信号被淬灭基团吸收,此时仪器不会检测到荧光信号。在PCR扩增时,模板DNA变性解链,Taqman探针特异性结合到目标基因处,而Taq酶具有5’-3’核酸外切酶活性,延伸至探针结合处,可将探针水解,使荧光报告基团和淬灭基团分离,从而检测到荧光信号,荧光信号的强度与扩增得到的dsDNA的浓度成正比。荧光信号被仪器检测并采集得到“扩增曲线”,通过荧光信号的强度反映出体系中dsDNA的浓度(见图1)。
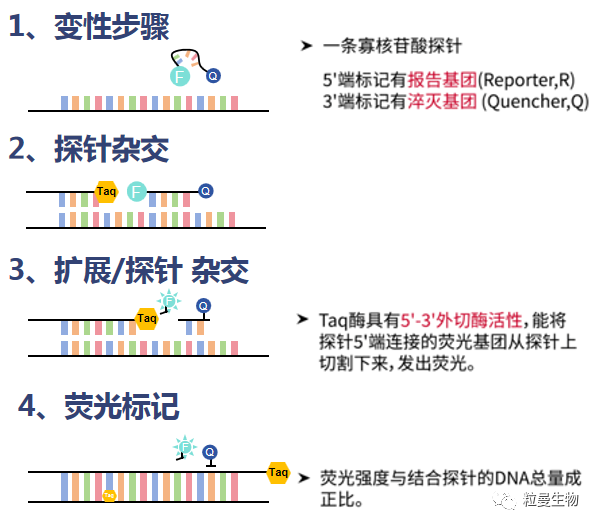
图1 qPCR 实验流程
2. qRT-PCR在评估KO细胞敲除效率中的应用
2.1 小的插入或缺失(indel)
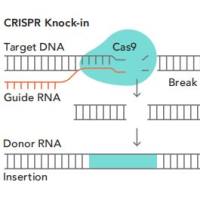
采用CRISPR/Cas9技术进行基因编辑时,向导RNA(guide RNA,gRNA)靶向目标基因,将在切割位点插入或删除少量碱基(indel),这些小的插入或缺失不会影响RNA转录,因此“编辑”的mRNA很可能仍然会产生(见图2)。针对这种情况,qRT-PCR是无法检测到目标基因发生了编辑。
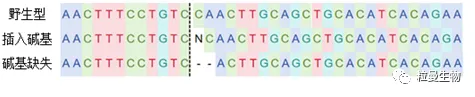
图2 插入或删除少量碱基图示
qRT-PCR可以检测和定量mRNA,但该技术不够灵敏,无法检测到目标基因小片段插入或删除,因此对于评估靶向单基因敲除产生的基因编辑,qRT-PCR技术不是100%准确的。
2.2 大片段缺失
如果在基因敲除时,产生了大片段的缺失,细胞转录过程中会产生截短的mRNA,在这种情况下,qPCR有可能检测到荧光信号的降低(见图3)。

图3 大片段缺失图示
2.3 产生终止密码子
基因被编辑后有可能产生终止密码子,引起翻译提前终止。产生的异常mRNA通过无义介导的RNA降解(NMD)机制[1](NMD是一个进化保守的RNA降解途径,其成为了细胞固有的一个有力限制逆转录病毒和正链RNA病毒的潜在机制)会被选择性清除。在这种情况下,RT-PCR可以检测到目标基因下调(见图4)。
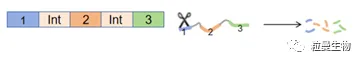
图4 NMD机制图示
总之,针对小的片段敲入,包括引入点突变(SNV)或密码子交换时, qRT-PCR方法可以在克隆验证和筛选过程中用于检测基因组DNA的编辑。但需要根据野生型序列和编辑后序列设计类似于单核苷酸多态性(SNP)qPCR分析的方法。在上述的情况下,Sanger测序、ddPCR或NGS仍然是比qRT-PCR更全面的了解不同亚群的编辑图谱,以及该区域可能发生的任何其他编辑。
3. 评估KO细胞敲除效率的推荐方案 粒曼团队建议通过Sanger一代测序或其他检测基因组序列(NGS、ddPCR)的技术来评估基因编辑效率,并通过WB(Western Blot)、蛋白质组学质谱鉴定等检测蛋白质表达水平的技术来确认蛋白质的缺失。(见图5)
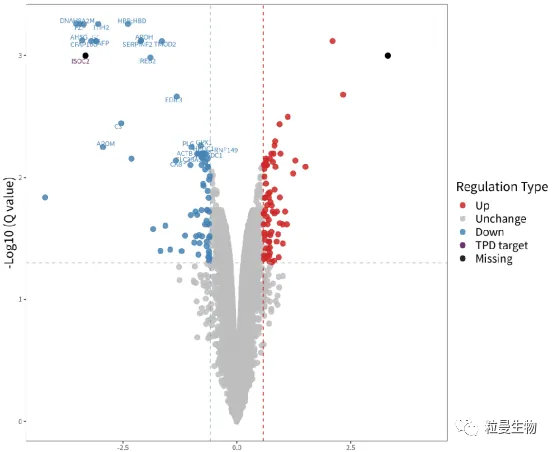
图5 通过蛋白质谱确认蛋白的缺失
在抗体特异性验证的方法中,基于CRISPR/Cas9的基因敲除 (KO) 验证的可信度最高。KO敲除验证采用的KO细胞系或裂解液是在细胞基因组上敲除编码靶蛋白的基因,从而消除该靶蛋白在细胞中的存在。测试抗体特异性时,野生型(WT)细胞中存在靶标蛋白与抗体结合,产生信号;KO细胞中不存在靶标蛋白,则不会产生信号。KO验证的方法对于确认抗体对靶标蛋白的特异性是非常有效的。(见图6)
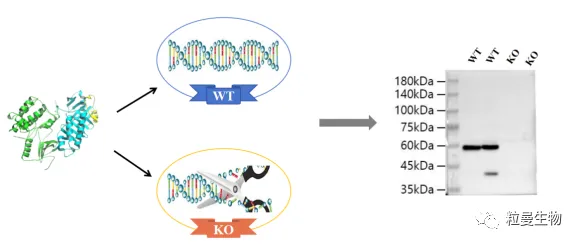
图6 通过KO验证过的抗体进行WB实验
粒曼团队强烈建议大家采购KO验证过的抗体进行WB等相关实验。粒曼提供高质量KO细胞裂解液产品,将帮助大家在实验前对手中的抗体进行特异性质控验证,避免实验误差和浪费宝贵的实验时间。
粒曼团队提供高通量基因敲除细胞系(池)构建服务,提供全面、科学的敲除效率评价方法,我们提供Sanger一代测序,Western Blot(KO验证过的抗体用于WB实验)以及基于质谱的蛋白质组学检测等多方位地评价KO效率。