



万千商家帮你免费找货
0 人在求购买到急需产品
- 详细信息
- 用户评价
- 文献和实验
- 技术资料
- 提供商:
深圳华大基因科技服务有限公司
- 服务名称:
真核转录组测序
- 规格:
200 µg/ml
转录组测序,对某一物种或特定细胞在某一功能状态下产生的mRNA进行高通量测序,既可以提供定量分析,检测基因表达水平差异,又可以提供结构分析,能发现稀有转录本,精确地识别可变剪切位点、基因融合等,而且不依赖于参考基因组。
技术优势
任意物种的全转录组分析:无需预先设计特异性探针,因此无需了解物种基因或基因组信息,能够直接对任何物种进行全面的转录组分析;
覆盖度高:数字化信号,直接测定几乎所有转录本片段的序列;
检测阈值宽:跨越6个数量级的宽检测阈值,从几个到数十万个拷贝精确计数;
分辨率高:可以检测基因家族中相似基因及可变剪接造成的单碱基差异;
检测范围广:从几个到数十万个拷贝精确计数,可同时鉴定及定量正常和稀有的转录本。
产品应用
医学上的应用方向: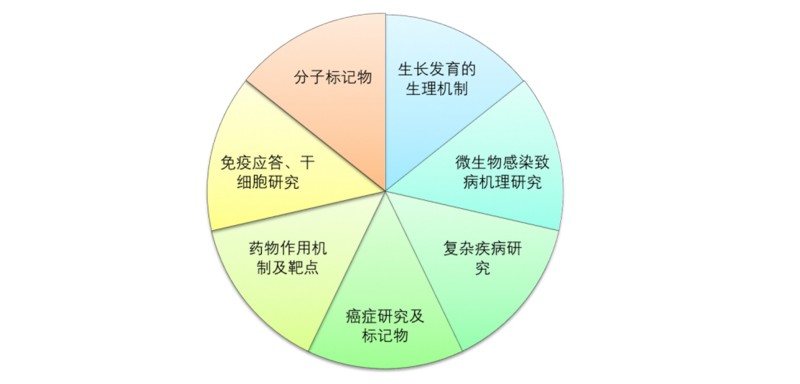
农学上的应用方向: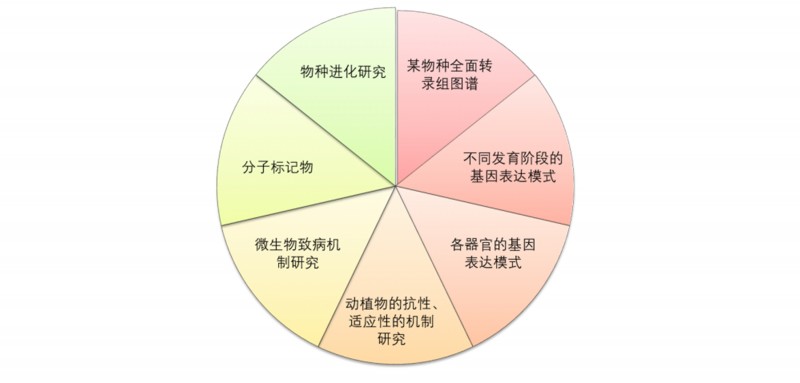
研究内容
一、无参考序列物种
标准信息分析:
1. 测序数据过滤;
2. 转录组de novo组装;
3. Unigene七大功能数据库注释;
4. Unigene的CDS预测;
5. Unigene的TF编码能力预测;
6. Unigene的SSR检测;
7. Unigene表达量计算(基因表达水平、PCA分析、基因表达水平箱线图和密度图、样品间的表达量韦恩图);
8. 时间序列分析;
9. 差异表达基因检测;
10. 差异表达基因聚类分析;
11. 差异表达基因GO功能分析;
12. 差异表达基因Pathway功能分析;
13. 差异基因蛋白互作分析;
14. 真菌致病基因预测 (真菌样本);
15. 植物抗病基因预测(植物样本)。
定制化信息分析:
1. 可结合客户的需求,协商确定定制化信息分析内容。
二、有参考序列物种
标准信息分析:
1. 基本数据统计① 去除接头序列、低质量序列得到reads信息,② 样品相关性,③ 表达量分布,④ RNA分类;
2. 参考基因组比对;
3. mRNA鉴定;
4. mRNA定量分析;
5. mRNA差异表达分析(样本间、组间);
6. mRNA表达/差异基因聚类;
7. mRNA差异基因GO分类、富集;
8. mRNA差异基因KEGG分类、富集;
9. mRNA结构分析:可变剪切分析;
10. mRNA结构分析:融合基因分析(仅限人)。
Dr. Tom信息分析:
一)数据库注释
1. 转录因子注释(AnimalTFDB/PlantTFDB);
2. GSEA分析;
3. Rfam、Pfam、Reactome、COG、EggNOG和InterPro数据库注释;
二)互作网络分析
1. 靶基因分析① miRNA-mRNA靶向关系分析,② lncRNA-mRNA靶向关系分析;
2. ceRNA互作网络分析;
3. 蛋白互作网络分析;
4. 共表达互作网络分析;
三)特色分析
1. 自定义标签和自有数据上传;
2. 外部数据库信息(TCGA、ARCHS4);
3. 关键驱动基因网络图分析;
4. 时间序列分析。
(*以上分析内容为部分物种可做。)
定制化信息分析 :
1. 可结合客户的需求,协商确定定制化信息分析内容
案例解析:
案例一 RAF样蛋白激酶介导一种高度保守且快速的生长素反应
文章题目:RAF-like protein kinases mediate a deeply conserved, rapid auxin response
发表期刊:Cell
影响因子:45.5
发表时间:2024年1月
发表单位:瓦赫宁根大学、查理大学、奥地利科学技术研究所、京都大学、东京理科大学
案例描述:该研究团队鉴定了一种对生长素快速全蛋白质组磷酸化的反应。这种反应发生在5种陆地植物和藻类物种上,并集中于一组核心的共同靶点。该研究鉴定出了这种生长素触发的跨物种磷酸化的中心介质是纤维肉瘤(RAF) 样蛋白激酶,遗传分析该激酶与生长素触发的蛋白质磷酸化和快速细胞反应联系起来,从而确定了绿色谱系中生长素快速反应的古老机制。
参考文献:Kuhn A, Roosjen M, Mutte S, Dubey SM, Carrillo Carrasco VP, Boeren S, Monzer A, Koehorst J, Kohchi T, Nishihama R, Fendrych M, Sprakel J, Friml J, Weijers D. RAF-like protein kinases mediate a deeply conserved, rapid auxin response. Cell. 2024 Jan
案例二 全球最大规模结直肠癌多组学研究
文章题目:Prognostic genome and transcriptome signatures in colorectal cancers
发表期刊:Nature
影响因子:50.5
发表时间:2024年8月
发表单位:中国科学院杭州医学研究所、瑞典乌普萨拉大学(Uppsala University)、华大生命科学研究院、华大基因智惠医学研究院
文章简述:该研究实现了迄今为止最大规模的结直肠癌基因组和转录组的综合分析,并将分子层面的发现与高质量的临床数据相结合,从而识别关键预后因子,这使这项研究有别于其他绝大多数癌症基因组学的研究。
研究结果:基于华大基因DNBSEQ测序平台,对瑞典U-CAN队列(Uppsala/Umeå Comprehensive Cancer Consortium)中的1,063例结直肠癌样本进行全基因组和转录组测序分析,鉴定得到了96个显著突变基因,其中,有24个为新发现的潜在驱动基因,并发现与WNT、EGFR和TGF-β通路相关的驱动基因和结直肠癌生存显著相关。利用突变时序分析,推导出9个与癌症发生早期事件相关的驱动突变基因和更倾向于在癌症发生后期阶段出现的突变基因。
这些发现为结直肠癌的早期检测和靶向治疗提供了临床研究策略,并揭示了与肿瘤后期侵袭和转移相关的重要分子变化。整合基因组和转录组数据的分子分型能得到更精细准确的患者预后分层,这对优化临床肿瘤分型、指导结直肠癌精准治疗具有重要意义,新构建的表达谱精细分型将在未来指导结直肠癌个体化诊疗中发挥重要作用。
参考文献:Nunes, L., Li, F., Wu, M. et al. Prognostic genome and transcriptome signatures in colorectal cancers. Nature (2024).
结果展示:
部分内容展示:
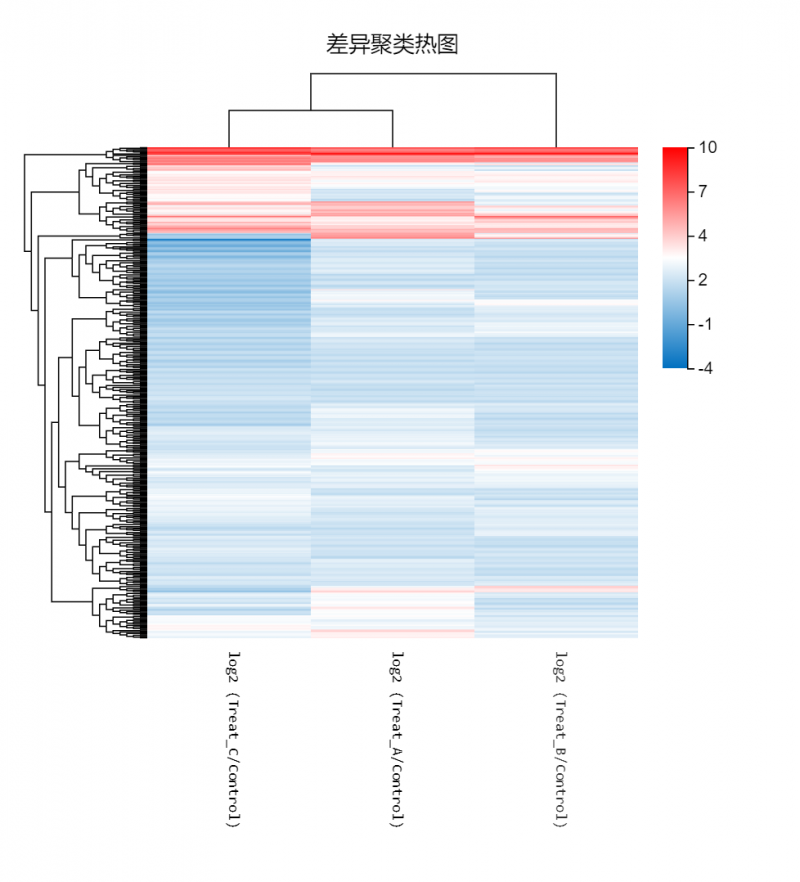
图1 差异聚类热图
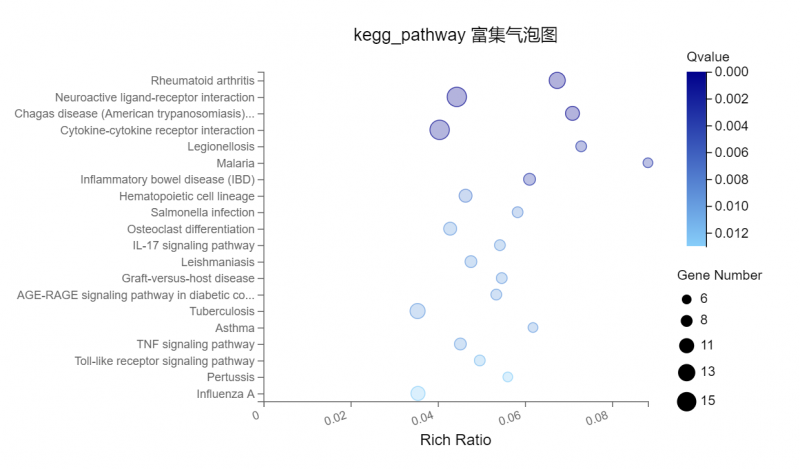
图2 功能富集或分类图

图3 蛋白互作网络
送样建议:
表1 真核转录组测序核酸样品判定标准
|
真核转录组(默认链特异性,非链特异和链特异性送样建议相同) |
|||||||
|
样本类型 |
总量 |
浓度 |
完整性 |
28S/18S |
基线和5S |
纯度 |
|
|
分析仪 检测 |
电泳检测 |
||||||
|
Total RNA (真菌) |
1 μg |
20-1,000 ng/μL |
RIN≥6.0 |
RNA条带清晰无弥散或拖尾 |
28S/18S≥1.0 |
基线平整,5S峰正常 |
无 DNA, 蛋白/盐离子等污染, 样本无色透明, 不粘稠 |
|
Total RNA (植物) |
400 ng |
10-1,000 ng/μL |
RIN≥6.0 |
28S/18S≥1.0 |
|||
|
Total RNA (其他动物) |
400 ng |
10-1,000 ng/μL |
RIN≥6.0 |
28S/18S≥1.0 |
|||
|
Total RNA (非全血人鼠) |
200 ng |
5-1,000 ng/μL |
RIN≥6.0 |
28S/18S≥1.0 |
|||
|
Total RNA (人鼠细胞系) |
200 ng |
5-1,000 ng/μL |
RIN≥7.0 |
28S/18S≥1.0 |
|||
|
Total RNA (昆虫) |
400 ng |
10-1,000 ng/μL |
RIN≥6.0 |
NA |
|||
表2 真核转录组测序组织样品判定标准
|
组织类型 |
真核转录组文库 |
|
新鲜培养细胞 (细胞数) |
≥2×105 cells |
|
新鲜动物组织干重 |
≥25 mg |
|
新鲜植物组织干重 |
嫩叶、嫩茎≥50mg 根、果实、种子、花等≥100mg |
|
菌体(细胞数或干重) |
≥5×106 cells 或 ≥20 mg |
|
全血-哺乳动物 |
≥1 mL 全血分离的白细胞 或≥1 mL PAXgene® Blood RNA Tube 收集的全血 或≥0.3mL 全血+3 倍体积 TRIzol裂解液 |
|
全血-非哺乳动物 |
≥0.1 mL 全血分离的白细胞 或≥0.1 mL 全血+6倍体积 TRIzol 裂解液 |
|
FFPE |
≥5片, 未染色, 组织面积≥100 mm2, 切片厚度5~10 μm |
风险提示:丁香通仅作为第三方平台,为商家信息发布提供平台空间。用户咨询产品时请注意保护个人信息及财产安全,合理判断,谨慎选购商品,商家和用户对交易行为负责。对于医疗器械类产品,请先查证核实企业经营资质和医疗器械产品注册证情况。
 用户评价
用户评价 暂无用户评价
暂无用户评价 文献和实验
文献和实验1、RAF-like protein kinases mediate a deeply conserved, rapid auxin response.Cell 2024
2、Prognostic genome and transcriptome signatures in colorectal cancers.Nature 2024
结合细胞在组织中分布的位置信息和转录组基因表达信息,了解细胞在空间背景下的异质性。
研究意义;而同样情况下RASGRP1的检测数据可能不能说明问题。 由此可知,设计的实验如果没有生物学重复,或者生物学重复的数量不够,就不能得到有统计意义的实验结果;获得的差异表达的基因很可能仅仅是少数个体差异的表现,并不能反映疾病或者某种特定生理状态的群体本质特征。 3.生物学重复设置几个合适? 您是不是有同样的问题:转录组测序是否必须进行生物学重复啊,是否要3个重复,是否可以用3个样品的RNA等量混合代替生物学重复,如果不重复能否发文章…..?一方面是有限的经费,一方面是编辑的质疑;实在很难
案例:应用全基因组测序和 RNA 测序来描绘常见的变异型免疫缺陷综合症(CVIDs)的基因图谱 背景:常见的变异型免疫缺陷综合症(CVIDs)是机体免疫应答反应中不能产生抗体的最主要原因。CVIDs 变异度很高,大概 5% 的病人是由基因改变引起的。 目的:利用 Illumina HiSeq2500 和 HTSeq 测序平台对 CVIDs 进行全基因组及转录水平的研究,揭示并绘制 CVIDs 的信号调控图谱。 结果:通过对 CVIDs 基因组和转录组进行测序和综合
 技术资料
技术资料暂无技术资料 索取技术资料

