

- ¥2980
- LMAI Bio
- LM-0849R-FITC
- 中国/美国/欧洲
- 2025年07月12日
- Flow-Cyt=1:50-200 IF=1:50-200
- Human, Mouse, Rat, Chicken, Pig, Cow,
- Human, Mouse, Rat, Chicken, Pig, Cow,

企业认证
相关产品推荐更多 >

万千商家帮你免费找货
0 人在求购买到急需产品
- 详细信息
- 文献和实验
- 技术资料
- 供应商:
上海联迈生物工程有限公司
- 库存:
大量
- 靶点:
详见说明书
- 级别:
1
- 目录编号:
LM-0849R-FITC
- 克隆性:
多克隆
- 抗原来源:
Rabbit
- 保质期:
1年
- 抗体英文名:
Anti-APOA1/FITC
- 抗体名:
Anti-APOA1/FITC
- 标记物:
FITC标记
- 宿主:
Human, Mouse, Rat, Chicken, Pig, Cow,
- 适应物种:
Human, Mouse, Rat, Chicken, Pig, Cow,
- 免疫原:
详见说明书
- 亚型:
IGg
- 形态:
粉末、液体、冻干粉
- 应用范围:
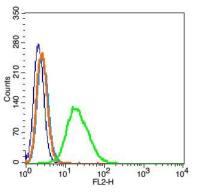
Flow-Cyt=1:50-200 IF=1:50-200
- 浓度:
1mg/ml
- 保存条件:
-20 °C
- 规格:
100ul
| 英文名称 | Anti-APOA1/FITC |
| 中文名称 | FITC标记的载脂蛋白A1抗体 |
| 别 名 | Apo-AI; ApoA I; ApoA-I; APOA1_HUMAN; Apolipoprotein A-I(1-242); Apolipoprotein A1; Apolipoprotein A 1; Apolipoprotein AI; Apolipoprotein A I; Brp14; Ltw1; Lvtw1; Sep1; Sep2. |
| 规格价格 | 100ul/2980元 购买 大包装/询价 |
| 说 明 书 | 100ul |
| 研究领域 | 心血管 免疫学 糖尿病 脂蛋白 |
| 抗体来源 | Rabbit |
| 克隆类型 | Polyclonal |
| 交叉反应 | Human, Mouse, Rat, Chicken, Pig, Cow, |
| 产品应用 | Flow-Cyt=1:50-200 IF=1:50-200 not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. |
| 分 子 量 | 28kDa |
| 性 状 | Lyophilized or Liquid |
| 浓 度 | 1mg/ml |
| 免 疫 原 | KLH conjugated synthetic peptide derived from human APOA1 |
| 亚 型 | IgG |
| 纯化方法 | affinity purified by Protein A |
| 储 存 液 | 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. |
| 保存条件 | Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C. |
| 产品介绍 | background: This gene encodes apolipoprotein A-I, which is the major protein component of high density lipoprotein (HDL) in plasma. The protein promotes cholesterol efflux from tissues to the liver for excretion, and it is a cofactor for lecithin cholesterolacyltransferase (LCAT) which is responsible for the formation of most plasma cholesteryl esters. This gene is closely linked with two other apolipoprotein genes on chromosome 11. Defects in this gene are associated with HDL deficiencies, including Tangier disease, and with systemic non-neuropathic amyloidosis. [provided by RefSeq, Jul 2008] Function: Participates in the reverse transport of cholesterol from tissues to the liver for excretion by promoting cholesterol efflux from tissues and by acting as a cofactor for the lecithin cholesterol acyltransferase (LCAT). As part of the SPAP complex, activates spermatozoa motility. Subunit: Interacts with APOA1BP and CLU. Component of a sperm activating protein complex (SPAP), consisting of APOA1, an immunoglobulin heavy chain, an immunoglobulin light chain and albumin. Interacts with NDRG1. Subcellular Location: Secreted. Tissue Specificity: Major protein of plasma HDL, also found in chylomicrons. Synthesized in the liver and small intestine. The oxidized form at Met-110 and Met-136 is increased in individuals with increased risk for coronary artery disease, such as in carrier of the eNOSa/b genotype and exposure to cigarette smoking. It is also present in increased levels in aortic lesions relative to native ApoA-I and increased levels are seen with increasing severity of disease. Post-translational modifications: Palmitoylated. Met-110 and Met-136 are oxidized to methionine sulfoxides. Phosphorylation sites are present in the extracelllular medium. DISEASE: Defects in APOA1 are a cause of high density lipoprotein deficiency type 2 (HDLD2) [MIM:604091]; also known as familial hypoalphalipoproteinemia (FHA). Inheritance is autosomal dominant. Defects in APOA1 are a cause of the low HDL levels observed in high density lipoprotein deficiency type 1 (HDLD1) [MIM:205400]; also known as analphalipoproteinemia or Tangier disease (TGD). HDLD1 is a recessive disorder characterized by the absence of plasma HDL, accumulation of cholesteryl esters, premature coronary artery disease, hepatosplenomegaly, recurrent peripheral neuropathy and progressive muscle wasting and weakness. In HDLD1 patients, ApoA-I fails to associate with HDL probably because of the faulty conversion of pro-ApoA-I molecules into mature chains, either due to a defect in the converting enzyme activity or a specific structural defect in Tangier ApoA-I. Note=A mutation in APOA1 is the cause of amyloid polyneuropathy-nephropathy Iowa type (AMYLIOWA); also known as amyloidosis van Allen type or familial amyloid polyneuropathy type III. AMYLIOWA is a hereditary generalized amyloidosis due to deposition of amyloid mainly constituted by apolipoprotein A1. The clinical picture is dominated by neuropathy in the early stages of the disease and nephropathy late in the course. Death is due in most cases to renal amyloidosis. Severe peptic ulcer disease can occurr in some and hearing loss is frequent. Cataracts is present in several, but vitreous opacities are not observed. Defects in APOA1 are a cause of amyloidosis type 8 (AMYL8) [MIM:105200]; also known as systemic non-neuropathic amyloidosis or Ostertag-type amyloidosis. AMYL8 is a hereditary generalized amyloidosis due to deposition of apolipoprotein A1, fibrinogen and lysozyme amyloids. Viscera are particularly affected. There is no involvement of the nervous system. Clinical features include renal amyloidosis resulting in nephrotic syndrome, arterial hypertension, hepatosplenomegaly, cholestasis, petechial skin rash. Similarity: Belongs to the apolipoprotein A1/A4/E family. Database links: Entrez Gene: 335 Human Omim: 107680 Human SwissProt: P02647 Human Unigene: 93194 Human Important Note: This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. |
风险提示:丁香通仅作为第三方平台,为商家信息发布提供平台空间。用户咨询产品时请注意保护个人信息及财产安全,合理判断,谨慎选购商品,商家和用户对交易行为负责。对于医疗器械类产品,请先查证核实企业经营资质和医疗器械产品注册证情况。
 文献和实验
文献和实验内腔可产生高强度的磁场,足以分离磁微粒标记的细胞,无需分离柱提供提供更高强度的磁场。由于磁微粒极小,不会影响靶细胞的流式分析结果,因此不需要去除。除了现有的定型产品外,客户可选择EasySep® PE、FITC或生物素分选系统与自己的PE、FITC或生物素—偶联的单抗相结合,来富集任何所需的细胞。如果您有自己的小鼠IgG1抗任何细胞的抗体, 也可以使用“Do-It-Yourself”试剂盒做成TAC,用来分选您想要的细胞。此外,还可以根据您的特殊需要,专门为您设计定做试剂盒。用于正选小鼠的细胞标记
举例。 表23-5 用基因作探针检测基因内的结构变化来直接分析遗传病 遗传病 探针 年代 抗凝血酶Ⅲ缺乏症 抗凝血酶Ⅲ基因 1983 α1抗胰蛋白酶缺乏症 合成的寡核苷酸 1983 动脉粥样硬化症 载脂蛋白A-基因 1983 糖尿病 胰岛素基因 1983 Ehlers-Danlos综合症 α(1)胶原蛋白基因 1983 生长激素缺乏症 生长激素基因 1981 乙型血友病 凝血因子ⅠⅩ基因 1983 遗传性胎儿血红蛋白持存症 β珠蛋白基因 1979,1983 HPRT
TFAR19(PDCD5)是由本研究室在国际上首先报导的一个拥有自己知识产权的人类新基因,前期的功能研究表明,它是促进细胞凋亡的增强剂。利用荧光素(FITC)标记的TFAR19单克隆抗体为探针,对细胞凋亡过程中TFAR19蛋白的表达水平及定位研究发现,凋亡早期TFAR19表达水平增高并出现快速核转位现象,伴随着细胞核形态学的变化,持续较长时间,在凋亡小体中仍然可见。同时我们发现,凋亡早期TFAR19蛋白的核转位早于磷脂酰丝氨酸(PS)外翻和细胞核DNA的片段化,提示TFAR19蛋白的核转
 技术资料
技术资料暂无技术资料 索取技术资料

