

- ¥1380 - 2200
- 康朗生物
- kl-4631R
- 中国/美国/德国
- 2025年12月23日
- IHC-P=1:400-800 IF=1:100-500
- Rabbit
- Human, Mouse, Rat, Dog,

企业认证
相关产品推荐更多 >


万千商家帮你免费找货
0 人在求购买到急需产品
- 详细信息
- 文献和实验
- 技术资料
- 供应商:
上海康朗生物科技有限公司
- 库存:
大量
- 目录编号:
kl-4631R
- 克隆性:
多克隆
- 抗原来源:
Rabbit
- 保质期:
12个月
- 抗体英文名:
Beta galactosidase antibody
- 抗体名:
β半乳糖苷酶抗体
- 宿主:
Rabbit
- 适应物种:
Human, Mouse, Rat, Dog,
- 免疫原:
KLH conjugated synthetic peptide derived from human Beta galactosidase (301-380aa):221-290/677
- 亚型:
IgG
- 形态:
冻干粉或液体
- 应用范围:
IHC-P=1:400-800 IF=1:100-500
- 浓度:
1mg/ml
- 保存条件:
-20 °C
- 规格:
100ul 200ul
| 中文名称 | β半乳糖苷酶抗体 |
| 别 名 | beta-Galactosidase; Beta D galactosidase; Beta gal; Beta galactosidase; EC 3.2.1.23; ECK0341; JW0335; lactase; lacZ; BGAL_HUMAN; Acid beta-galactosidase; Lactase; Elastin receptor 1. β-Galactosidase. |
| 规格价格 | 100ul/1380元 购买 200ul/2200元 购买 大包装/询价 |
| 说 明 书 | 100ul 200ul |
| 研究领域 | 免疫学 细胞类型标志物 |
| 抗体来源 | Rabbit |
| 克隆类型 | Polyclonal |
| 交叉反应 | Human, Mouse, Rat, Dog, |
| 产品应用 | IHC-P=1:400-800 IF=1:100-500 (石蜡切片需做抗原修复) not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. |
| 分 子 量 | 71kDa |
| 细胞定位 | 细胞浆 |
| 性 状 | Lyophilized or Liquid |
| 浓 度 | 1mg/ml |
| 免 疫 原 | KLH conjugated synthetic peptide derived from human Beta galactosidase (301-380aa):221-290/677 |
| 亚 型 | IgG |
| 纯化方法 | affinity purified by Protein A |
| 储 存 液 | 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. |
| 保存条件 | Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C. |
| PubMed | PubMed |
| 产品介绍 | background: Beta galactosidase is coded by a gene (lac z) in the lac operon of Escherichia coli. It is a metalloenzyme that splits lactose into glucose and galactose. It hydrolyzes terminal, non-reducing beta-D-galactose residues in beta-D-galactosides. Activation by cations seems to be substrate dependent. K+, Na+, NH4+, Rb+, Cs+ and Mn++ all activate enzyme activity based upon the substrate used. Function: Cleaves beta-linked terminal galactosyl residues from gangliosides, glycoproteins, and glycosaminoglycans. Isoform 2 has no beta-galactosidase catalytic activity, but plays functional roles in the formation of extracellular elastic fibers (elastogenesis) and in the development of connective tissue. Seems to be identical to the elastin-binding protein (EBP), a major component of the non-integrin cell surface receptor expressed on fibroblasts, smooth muscle cells, chondroblasts, leukocytes, and certain cancer cell types. In elastin producing cells, associates with tropoelastin intracellularly and functions as a recycling molecular chaperone which facilitates the secretions of tropoelastin and its assembly into elastic fibers. Subcellular Location: Isoform 1: Lysosome. Isoform 2: Cytoplasm, perinuclear region. Note=Localized to the perinuclear area of the cytoplasm but not to lysosomes. DISEASE: Defects in GLB1 are the cause of GM1-gangliosidosis type 1 (GM1G1) [MIM:230500]; also known as infantile GM1-gangliosidosis. GM1-gangliosidosis is an autosomal recessive lysosomal storage disease marked by the accumulation of GM1 gangliosides, glycoproteins and keratan sulfate primarily in neurons of the central nervous system. GM1G1 is characterized by onset within the irst three months of life, central nervous system degeneration, coarse facial features, hepatosplenomegaly, skeletal dysmorphology reminiscent of Hurler syndrome, and rapidly progressive psychomotor deterioration. Urinary oligosaccharide levels are high. It leads to death usually between the first and second year of life. Defects in GLB1 are the cause of GM1-gangliosidosis type 2 (GM1G2) [MIM:230600]; also known as late infantile/juvenile GM1-gangliosidosis. GM1G2 is characterized by onset between ages 1 and 5. The main symptom is locomotor ataxia, ultimately leading to a state of decerebration with epileptic seizures. Patients do not display the skeletal changes associated with the infantile form, but they nonetheless excrete elevated amounts of beta-linked galactose-terminal oligosaccharides. Inheritance is autosomal recessive. Defects in GLB1 are the cause of GM1-gangliosidosis type 3 (GM1G3) [MIM:230650]; also known as adult or chronic GM1-gangliosidosis. GM1G3 is characterized by a variable phenotype. Patients show mild skeletal abnormalities, dysarthria, gait disturbance, dystonia and visual impairment. Visceromegaly is absent. Intellectual deficit can initially be mild or absent but progresses over time. Inheritance is autosomal recessive. Defects in GLB1 are the cause of mucopolysaccharidosis type 4B (MPS4B) [MIM:253010]; also known as Morquio syndrome B. MPS4B is a form of mucopolysaccharidosis type 4, an autosomal recessive lysosomal storage disease characterized by intracellular accumulation of keratan sulfate and chondroitin-6-sulfate. Key clinical features include short stature, skeletal dysplasia, dental anomalies, and corneal clouding. Intelligence is normal and there is no direct central nervous system involvement, although the skeletal changes may result in neurologic complications. There is variable severity, but patients with the severe phenotype usually do not survive past the second or third decade of life. Similarity: Belongs to the glycosyl hydrolase 35 family. SWISS: P16278 Gene ID: 2720 Database links:
Important Note: This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. |
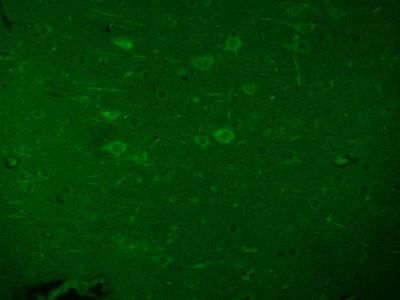
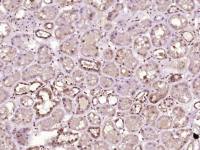
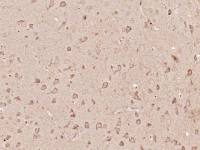
| 产品图片 | 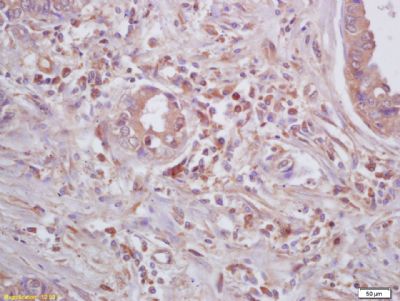 Tissue/cell: human lung carcinoma; 4% Paraformaldehyde-fixed and paraffin-embedded; Antigen retrieval: citrate buffer ( 0.01M, pH 6.0 ), Boiling bathing for 15min; Block endogenous peroxidase by 3% Hydrogen peroxide for 30min; Blocking buffer (normal goat serum,C-0005) at 37℃ for 20 min; Incubation: Anti-Beta galactosidase Polyclonal Antibody, Unconjugated(bs-4631R) 1:200, overnight at 4°C, followed by conjugation to the secondary antibody(SP-0023) and DAB(C-0010) staining 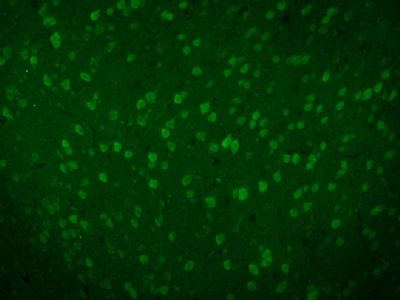 Paraformaldehyde-fixed, paraffin embedded (Mouse brain); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (Beta galactosidase) Polyclonal Antibody, Unconjugated (bs-4631R) at 1:200 overnight at 4°C, followed by a conjugated Goat Anti-Rabbit IgG antibody (bs-0295G-AF488) for 90 minutes, and DAPI for nuclei staining. 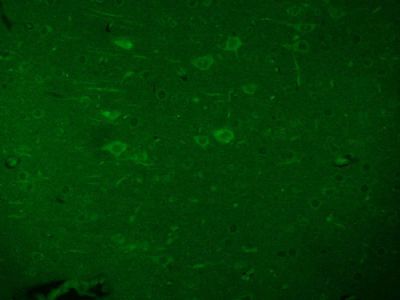 Paraformaldehyde-fixed, paraffin embedded (Rat brain); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (Beta galactosidase) Polyclonal Antibody, Unconjugated (bs-4631R) at 1:200 overnight at 4°C, followed by a conjugated Goat Anti-Rabbit IgG antibody (bs-0295G-AF488) for 90 minutes, and DAPI for nuclei staining. |
风险提示:丁香通仅作为第三方平台,为商家信息发布提供平台空间。用户咨询产品时请注意保护个人信息及财产安全,合理判断,谨慎选购商品,商家和用户对交易行为负责。对于医疗器械类产品,请先查证核实企业经营资质和医疗器械产品注册证情况。
 文献和实验
文献和实验Anticardiolipin Antibody and Anti-beta 2 Glycoprotein I Antibody Assays
) antibodies and anti-beta 2 glycoprotein I (β2-GPI) antibodies. Other autoantibodies, such as those directed against anionic phospholipids, can also be assayed; however they are not of clinical significance. Participation in a quality assurance
Cell Staining for Senescence-Associated beta-Galactosidase (SA-beta-Gal) Activity
Description Cell Staining for Senescence-Associated â-Galactosidase (SA-â-Gal) Activity Procedure 1. Carefully remove the growth media from the cell culture. 2. Add an equal volume of PBS and gently mix the PBS solution
Beta-Galactosidase Staining in the Skeleton
The lacZ gene, encoding for the β-galactosidase enzyme, is widely used as a reporter gene in bone biology due to the ease of visualization in situ on whole-mount or on tissue sections. In this protocol we provide detailed methods
 技术资料
技术资料暂无技术资料 索取技术资料
