


- 询价
- 原核转录组测序
- 全国
- 2026年06月05日

企业认证





万千商家帮你免费找货
0 人在求购买到急需产品
- 详细信息
- 文献和实验
- 技术资料
- 提供商:
北京诺禾致源科技股份有限公司
- 服务名称:
转录组测序测序
高质量测序准确分析mRNA信息
原核转录组测序是基于Illumina测序平台,构建链特异性文库,研究原核生物在某个时期或者在某种环境条件下转录出来的所有mRNA。应用方向
-
环境影响
分析环境变化对微生物的生物学功能 -
胁迫研究
研究胁迫环境下微生物的机体响应机制 -
差异性状研究
寻找不同处理导致差异性状或表型相关联的主要功能基因
诺禾优势
1. 样本起始量低
RNA 1 ug/建库(原核链特建库)
2.周期快,质量好,稳定性高
诺禾原核转录组测序基于高通量测序,分析物种在特定条件下转录所得所有mRNA信息,系统全面的分析mRNA在结构和表达水平的变化,为原核生物的深入研究奠定基础。

3.方案设计专业,质控严格
诺禾原核转录组测序采用去rRNA原核链特异性的建库方式;严格要求测序数据质量:Q20>90%,Q30>85%。从材料选取,建库测序,到数据分析, 每一步都经过科学、缜密的设计,以保障高质量研究成果。
同时诺禾致源高性能计算平台(High Performance Computing,HPC)采用DELL计算节点和Isilon存储的高效组合,实现快速稳定的测序数据分析及交付。随着公司业务的发展,高性能计算平台将 会持续更新并扩容,以保证高效的数据处理和安全的数据存储。
4.项目文章以及物种经验丰富
诺禾致源已经成功对大肠杆菌、鲍曼不动杆菌、副溶血弧菌、荧光假单胞菌、牙龈卟啉单胞菌、水稻黄单胞杆菌、水稻条斑病菌、乳酸菌、链霉菌、发状念珠藻、小球藻、拟柱胞藻、嗜盐古菌 等几百个物种进行原核转录组测序分析,并在该领域研究的权威期刊Advanced science等杂志发表高水平文章多篇。

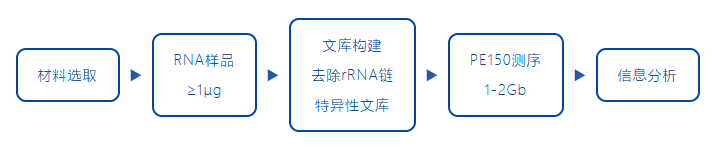
信息分析
诺禾致源原核转录组测序项目,除了基因表达以及差异基因之外,还包括多项基因结构水平的分析。表达水平分析包括基因表达量分析、相关性分析、差异基因及其富集分析、WGCNA分析等, 结构水平包括UTR分析、变异位点分析、新转录本预测等等。| 原核转录组 | 分析内容 | 解决问题 |
|---|---|---|
| 标准分析 | 测序数据质量评估 | 过滤掉低质量数据,保证数据质量 |
| 与参考基因组比对 | 比对率分析 | |
| 基因表达水平分析 | 分析基本的表达量 | |
| 样本相关性分析 | 分析样本间的相关性大小 | |
| 差异基因分析 | 寻找不同比较组合间的差异基因 | |
| GO/KEGG富集分析 | 差异基因的GO,KEGG富集分析 | |
| GSEA分析 | 基因集富集分析 | |
| 蛋白互作网络分析 | 差异基因蛋白互作网络的分析 | |
| WGCNA基因共表达网络分析 | 分析模块与样本间的相关性,寻找关键模块和hub基因 | |
| 高级分析 | SNP/Indel分析 | 分析变异位点 |
| 新转录本预测 | 挖掘该物种新的基因或转录本 | |
| 基因结构分析 | 对操纵子、转录起始位点和转录终止位点进行预测,然后进行启动子预测。 | |
| UTR注释和相关分析 | 5’UTR SD序列预测和3’UTR 终止子预测 | |
| 反义转录本预测 | 鉴定其反义转录本在基因组上的位置、种类以及数量等。 | |
| sRNA分析 | 对候选的sRNA分别进行二级结构预测和靶基因预测 |
送样建议
| 组织类型 | 送样建议 |
|---|---|
| total RN | 浓度:≥ 50ng/μL 体积:20μL ≤ V ≤ 120μL 总量:≥ 1μg |
| 菌体/菌丝 | ≥ 1*107个 ≥ 0.5g |
常见问题
1.物种没有参考基因组,可以做原核转录组测序吗?
不可以。原核转录组测序要求有参考基因组,因为原核生物的mRNA一般为多顺反子,直接拼接效果较差。
2.原核转录组测序的深度和覆盖度是多少?
一般组装基因组测序的项目才会涉及到测序深度和覆盖度,基因组测序项目中测序深度是指测序得到的总碱基数与待测基因组大小的比值。假设一个基因大小为2M,测序深度为10X, 那么获得的总数据量为20M。覆盖度是指测序获得的序列占整个基因组的比例。由于基因组中的高GC、重复序列等复杂结构的存在,测序最终拼接组装获得的序列往往无法覆盖有所的 区域,这部分没有获得的区域就称为Gap。
转录组测序(RNA-seq)一般不涉及深度和覆盖度两个概念,大多考虑的是样本测序数据量,和mapping率指标。一般使用以下的计算方式计算:深度= 数据量大小 / 参考基因组大小; 覆盖度的计算可参考:github.com/shiquan/bamdst。原核转录组测序通常建议测序数据量1-2G即可。
3.能否提取部分基因来做差异分析?
不能。差异分析是基于整体来做的。差异分析软件的作者推荐用全部readcount进行差异分析,若使用部分基因做分析,会毁坏掉数据整体的特点,如测序深度、reads分布特征。所以不推荐老师抽取部分来做差异分析。其实这个问题不只是针对原核转录组测序,其他产品中的差异分析也是同样的回答。
4.如果想从转录水平研究牛的瘤胃微生物,该怎么做?
可以做宏转录组分析。像瘤胃微生物(或肠道微生物)种类比较多,且难以分离培养,甚至有的微生物很难在体外培养,不建议做单个物种的原核转录组测序,可以直接对整个“环境” 中的微生物进行RNA-seq。宏转录组项目可以从整体水平研究某一特定环境,特定时期群体生命全部基因组转录情况以及转录调控规律,可以很好地避开微生物分离培养困难的问题。
风险提示:丁香通仅作为第三方平台,为商家信息发布提供平台空间。用户咨询产品时请注意保护个人信息及财产安全,合理判断,谨慎选购商品,商家和用户对交易行为负责。对于医疗器械类产品,请先查证核实企业经营资质和医疗器械产品注册证情况。
 文献和实验
文献和实验[1] Qiao, Qin et al. “The genome and transcriptome of Trichormus sp. NMC-1: insights into adaptation to extreme environments on the Qinghai-Tibet Plateau.” Scientific reports vol. 6 29404. 6 Jul. 2016, doi:10.1038/srep29404
结合细胞在组织中分布的位置信息和转录组基因表达信息,了解细胞在空间背景下的异质性。
于将3个样本的基因表达量取了平均值,其实就是相当于取了一个样本,由此得到的差异基因同样不可信,不能反应群体生物学现象。 4.生物学重复分析结果展示 以公司做项目的经验来看,原核生物以及真菌生物学重复的效果>植物>动物,这是由于动植物个体差异较大所导致。所以动植物在选取生物学重复时,应按照严格的筛选条件进行取样,方可得到理想结果。 案例二:真菌的转录组测序 实验设计:处理组与对照组(n=3) 实验结果:如下图所示,重复样本间的相关系数r>0.9 案例三:植物转录组测序 实验
案例:应用全基因组测序和 RNA 测序来描绘常见的变异型免疫缺陷综合症(CVIDs)的基因图谱 背景:常见的变异型免疫缺陷综合症(CVIDs)是机体免疫应答反应中不能产生抗体的最主要原因。CVIDs 变异度很高,大概 5% 的病人是由基因改变引起的。 目的:利用 Illumina HiSeq2500 和 HTSeq 测序平台对 CVIDs 进行全基因组及转录水平的研究,揭示并绘制 CVIDs 的信号调控图谱。 结果:通过对 CVIDs 基因组和转录组进行测序和综合
 技术资料
技术资料暂无技术资料 索取技术资料

