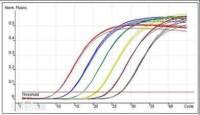
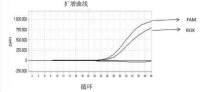
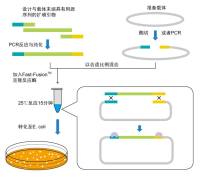
7 年
手机商铺
入驻年限:7 年
韩丽君
上海 青浦区
试剂、抗体、细胞库 / 细胞培养、ELISA 试剂盒、耗材
代理商 经销商 生产厂商 科研机构
哈维氏弧菌核酸检测试剂盒(恒温荧光法)售后服务
品牌:西格
询价
对虾传染性肌肉坏死病毒(IMNV)RNA核酸检测试剂盒(恒温荧光法)售后服务
罗氏沼虾诺达病毒(MrNV)RNA核酸检测试剂盒(恒温荧光法)多少钱
对虾黄头病毒(YHV)RNA核酸检测试剂盒(恒温荧光法)价格
对虾杆状病毒(BP)核酸检测试剂盒(恒温荧光法)说明书
19 人阅读发布时间:2026-07-22 11:40
17 人阅读发布时间:2026-07-21 15:47
16 人阅读发布时间:2026-07-17 11:02
13 人阅读发布时间:2026-07-15 11:46
20 人阅读发布时间:2026-07-13 15:09
28 人阅读发布时间:2026-07-08 13:18
37 人阅读发布时间:2026-07-06 14:36
25 人阅读发布时间:2026-07-03 09:22
37 人阅读发布时间:2026-06-29 13:35
24 人阅读发布时间:2026-06-24 10:58
34 人阅读发布时间:2026-06-22 16:53
70 人阅读发布时间:2026-06-18 14:25
70 人阅读发布时间:2026-06-17 14:52
21 人阅读发布时间:2026-06-15 09:12
46 人阅读发布时间:2026-06-12 10:52
48 人阅读发布时间:2026-06-10 10:30
68 人阅读发布时间:2026-06-08 14:04
57 人阅读发布时间:2026-06-03 13:49
101 人阅读发布时间:2026-06-01 14:02
81 人阅读发布时间:2026-05-29 10:44
在线沟通
我的询价