

基础知识

提高酶稳定性是改造酶在高温等极端条件下行使其活性理想的设计步骤,同时也能降低酶对蛋白酶剪切的敏感性。由于这些优点,研究者已经发明了很多不同的方法和技术改造蛋白质,但到目前为止这些方法的效果很有限。这里,我们介绍包括末端截切、随机突变和断裂、重组、再延长,然后在生理温度下筛选,以期获得稳定性提高的酶活突变体。以 TEM-1β-内酰胺酶为模型蛋白,通过三轮酶的定向进化,包括随机突变、DNA 混编以及在 37℃ 下的筛选,得到了其体内氨苄抗性与野生型相当的有删截的突变体。动力学研究表明,筛选到的突变体与其野生型相比有显著的热稳定性。这种方法非常有效,进化所产生的酶能够在比野生型的最适温度高 20°C 的条件下,保持其最高的酶活反应。因此在生理温度下通过末端截切和接下来作为补偿的定向进化来干扰结构,是提高蛋白质热稳定性的快速、高效的方法,避免了在高温下筛选的需要。

蛋白质工程领域主要涉及关于设计新的酶活性或折叠,以及理解蛋白质正确折叠和稳定的最基本的序列决定因素。迄今,已有巨大的精力投入到设计构建多肽文库方法的研究中。最常用的方法是用来筛选具有高亲和力的特异性结合蛋白的噬菌体展示技术。本章讲述了一种可替代噬菌体展示的方法,该方法是基于二氢叶酸还原酶(DHFR)蛋白质片段互补检测(PCA)建立起来的,可以完全用于体内实验。我们以筛选 raf 蛋白的 ras 结合结构域(ras-binding domain,RBD)正确折叠且能于 ras 结合所需的简并序列为例,讲述 DHFR PCA 的应用。此筛选系统通过重复的竞争实验,富集文库中表现最好的序列,而无需将体外实验所必需的文库筛选和扩增步骤分离。而且,可以在 96 孔板中通过三乙酸基氮( nitrilotriacetic acd,NiNTA)亲和层析直接快速处理被选中的克隆。此方法特别适用于设计和筛选研究序列折叠和结合决定因素的文库。另外,将该方法略加修改,还可以应用于文库间的筛选,从而可以使相互作用的蛋白质实现共同进化。
分隔式自我复制(CSR)是蛋白酶,特别是聚合酶,定向进化的一种新方法。在 CSR 最简单形式中,它包括一个简单的环路反馈,即聚合酶只复制编码自身的基因 (自我复制)。自我复制发生在一个热稳定的油包水乳化液形成的离散、分离、没有交互的空间中。分隔保证了表型和基因型之间的正确联系(也就是说,分隔保证了聚合酶只复制编码自身的基因而与空间外的成分没有关系)。这样,聚合酶特性的改进直接转化到编码该聚合酶的基因信息的扩增上。CSR 被证明在聚合酶的定向进化上是一个很有用的方法。在本章,我们描述了一些有用的 CSR 的实验方法,这些方法成功地利用一个 Tag 聚合酶随机突变库,使 Poll(Tag) 聚合酶性质得到了改变,如增加了热稳定性以及对潜在抑制剂(肝素)的抵抗能力。
在这里我们介绍一种在体外将基因型和表现型相联系的新策略,用来选择功能蛋白质。这个策略里,蓖麻蛋白的 A 链在翻译时被激活,因此不需要去掉终止密码子,就能形成核糖体、信使 mRNA 和翻译出蛋白质的稳定复合物。这个技术不需要转染、化学合成、连接,也不需要去掉终止密码子。因此,我们的新型核糖体失活展示系统能够为体外蛋白质的进化提供一个可靠的、简单的、完全的筛选系统,而不损失种群库,这样将随机和适度选择策略综合应用的方法,可以预测进化方向。
本章描述了改进融合蛋白在 M13 噬菌体颗粒表面展示水平的一个方法。向 M13 衣壳锚定蛋白引入突变后,蛋白质展示水平约能增加两个数量级。这里讲述改进蛋白质展示水平的噬菌体展示库的设计、构建以及筛选方法。
蛋白酶生化性质的改善可以通过对该酶基因的错掺突变和 DNA 混编方法实现。混编技术可用于同一基因的一组突变体,或者对相关家族基因的片段进行新的组合,产生嵌合突变基因产物。但该方法有一个缺点,就是在混编重组过程中,突变库中会存留大量并未发生重组的(“亲本”)片段。现在,我们可以通过设计简并引物,利用简并寡核苷酸基因改组方法来提髙发生重组基因的产量,减少未重组“亲本”基因的产量。在实验过程中,该方法既可避免混编前使用核酸内切酶将基因切成片段,又对基因特定片段进行随机突变。这里我们详细描述了这种方法如何应用于不同 GC 含量的 β-木聚糖酶家族基因。
利用 DNA 混编模仿自然进化过程是优化 DNA 和蛋白质性质的常用方法。这里我们介绍这类方法的一个新进展,即利用标准的聚合酶链反应(PCR)放大基因文库时与其他 4 种标准 dNTP—起掺入 dUTP 作为确定 DNA 碎裂位点的交换核苷酸。掺入的尿嘧啶碱基可用尿嘧啶-DNA-糖基化酶切除,随后 DNA 主链用哌啶切开。这个寡聚核苷酸库的重组装由内引物延伸步骤及高保真聚合酶来增加产率,最后由 PCR 扩增。变性聚丙烯酰胺尿素凝胶电泳表明这个方法可以产生大小可调的 DNA 片段,大小范围取决于 dUTP :dTTP 比率。对于一个模式蛋白,氯霉素乙酰转移酶I( CAT ),利用包含 33% dUTP 的 PCR 产生的混编基因库测序显示了大约 0.1% 的低突变率,并且,在没有进行片段分离的情况下,平均 亲本片段大小为 86 个碱基。因此,核苷酸交换和剪切技术(NExT) DNA 混编可重复性好并且容易实行,使之优于相应同类技术。另外,NExT 碎裂的结果可以利用计算机软件,NexTProg 预测出来。
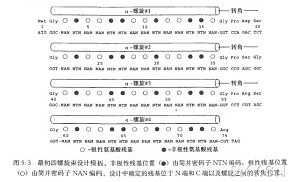
设计具有很好折叠的大规模新型蛋白质库是最终能够产生具有工业及医学应用新结构及功能的蛋白质的有效途径。本章介绍一个极性/非极性氨基酸“二元组图”的蛋白质库设计方法,此方法整合了合理化设计和重组多样性。“二元组图”基于的前提是极性/非极性氨基酸残基的适当组合可以指导多肽链折叠成具有双亲性质的二级结构,此类二级结构可能“退火”形成所需的高级结构。设计的二元组图利用蛋白质二级结构中本身具有的周期性,使得每一个极性和非极性位点的侧链可以产生的组合变化。本章概述了如何利用二元组图法设计新型蛋白质库。

在设计蛋白质库进行筛选时,在能够进行筛选的物理极限内我们必须尽一切可能产生出蛋白质变体的多样性。本章的目标是将概率的语言引入蛋白质工程实验中,进而来回答一些常见的问题。例如,我们怎样才能最有效地设计蛋白质库?我们最终从一个多样性完备的理论蛋白质库(简称理论库)中取样的比率是多少?错过理论库中一个个体的概率是多少?我们选择的变体中的突变是否具有统计意义,或仅仅是随机变异的产物?在整个蛋白质工程实验过程中这些计算标准可以使我们更好地设计和评价我们得出的蛋白质变体库。
位点特异核酸内切酶涉及核酸生物化学的许多方面。限制酶及相关酶已成为作用于 DNA 的酶的典范。通过理性蛋白设计,投入了无数的精力试图改变其特异性。但是,从整体上说,成功很少,大概是由于其识别能力高度冗余,并且识别和催化紧密地耦合。本章描述少数几个成功改变特异性例子之一,即转换错配修复核酸酶 MutH,当被 MetS 和 MutL 刺激的时候,此酶在半甲基化位点 d ( GATC) 切开一缺口——转化为切割全甲基化以及半甲基化和未甲基化 DNA 的变体。本章将描述此设计项目中所涉及的各种步骤,从对结构的分析和对负责感知甲基化状态的候选氨基酸残基的鉴别,到产生和鉴定针对半甲基化 d (GATC)位点具有不同特异性的 MutH 变体。
我们发展了用人纤连蛋白的第 10 个纤连蛋白类型Ⅲ结构域(FNfnlO)作为框架以显示用于与其他分子结合的多重表面链套的用法。我们把具有新的结合功能的 FNfnlO 变体称为“独体(孤体、单小体)”(FNfnlO 是一个由 94 个氨基酸组成的小蛋白质,它具有近似免疫球蛋白折叠的β三明治结构)。它是局度稳定的,没有二硫键或金属离子,它在细菌中高水平地表达为正确的折叠形式。这些期望的物理性质,使得 FNfnlO 框架与其他任何展示技术,事实上都可以比较。本章描述实物库构建和筛选以及独体的生产。
在 DNA 结合模体中,(Cys)2(His)2 类型的锌指模体具有大的操控潜力。为新的 DNA 结合蛋白设计,锌指模体提供了有吸引力的框架。特别是为产生新的、具有全新的 DNA 结合特性的,如长 DNA 链识别、DNA 弯折和 AT 富集序列识别——人造锌指蛋白,基于结构的设计技巧是特别有价值的。在此,基于最新的实验结果,描述为设计多重锌指蛋白以用于目标 DNA 序列识别、DNA 弯折锌指蛋白、(His)4类型的锌指蛋白和 AT 识别锌指蛋白的新技巧。
钙调素(CaM) 是普遍分布的蛋白质,参与钙介入的信号转导。当 Ca2+ 流入时,CaM 获得强亲和力,结合各种带有一个或多个CaM 识别序列的胞内蛋白,启动或终止 Ca2+ 调控的信号串联。通过对 Ca2+-CaM 复合物的核磁共振和晶体学结构研究,我们已得到对 CaM 目标识别机制的深入了解。一个最直接的应用就是,基于蛋白质的 Ca2+ 传感器。它使用了 CaM 复合物和绿色荧光蛋白,原先称为“chameleon”(钙变色子,英文原意为 变色龙——译者)。钙变色子的主要优点是,它们可以在单个细胞中表达,并以特定组织或细胞为目标,测量局部 Ca2+ 的变化。本章描述有关的方法,包括钙变色子的克隆、鉴定它们的生物化学和生物物理特征,以及用数字荧光显微镜在单个细胞中使它们成像。
虽然表观上简单,盘绕螺旋(coiled coil)模体是高度专一的,并在理解三级结构及其形成方面具有重要意义。最常观察到的盘绕螺旋形态——平行二聚态,其一般的结构类型仍有待全面的描述。尽管如此,其结构已呈现出在某些特定位置需要某些特定类型氨基酸的严格规则。本章我们基于现有盘绕螺旋结构,就应用这些规则到要设计或优化的盘绕螺旋结构来讨论这个系统。基于这些规则之上的理解和扩展,对这些模体的应用是关键的。因为,这些模体实际上在每个细胞过程中都起关键作用,它们通过屏蔽其他非天然的或不规则的蛋白质(如在肿瘤形成中跟盘绕螺旋结构域结合),作为药物投递试剂。我们讨论其结构中的 a 和 d 的 “疏水”核心位置以及 e 和 g 的“静电”边缘位置,在引导寡聚化和配对化的特异性作用。同样被讨论的还有那些在维持 a 螺旋倾向性、螺旋可溶性和二聚体稳定性上与 b、c 和 f 位有协同作用的位置。
氨基酸类似物的掺入越来越有用。非天然氨基酸的定点掺入,使得运用化学生物学对特定蛋白质的研究和应用成为可能。但是,非天然氨基酸的整体掺入也检验着蛋白质组和基因编码假定。例如,有机体对非天然氨基酸的适应可能会导致新的基因编码。为了理解和定量化这样的掺入引起的变化,需理解微生物学和蛋白 质组对非天然氨基酸掺入的反应。此文描述了在蛋白质组范围鉴定这些掺入效应的规程。
计算方法一直在蛋白质设计中发挥重要作用。本工作主要集中在搜索蛋白质序列空间,以找到一条或数条与已知结构和功能相容的蛋白质序列。在期望的功能和结构限制下,概率性计算方法为所容许的氨基酸变化范围提供信息。这样的方法可用于指导 建立蛋白质的单个序列或组合库。
无论是保存还是运输,请避免反复冻融。反复冻融,冰晶会破坏抗体和重组蛋白的空间结构,导致蛋白变性形成多聚体和重组蛋白构象改变,从而降低抗体的结合能力,也加快了抗体球蛋白和重组蛋白的降解速度。抗体和重组蛋白保存得当与否,直接决定了抗体和重组蛋白的活性使用效果,如果保存得当,Immune tech(苏州杰恩生物)的抗体和重组蛋白活性大部分都可以维持数年。1. 收到抗体和重组蛋白后的操作收到抗体和重组蛋白后(大部分抗体和重组蛋 ...
xiaonichi 我现在在做一个蛋白,命名为fkbp38,按道理应该是38kd。因为同家族的fkbp12为108aa,约12kd但现在在ncbi上找到的序列,人的有413个aa,显示的分子量为44-45kd左右这是啥原因呢?xianyuanshan 楼主找到的是这个吗?—— NP_036313peptidyl-prolyl cis-trans isomerase FKBP8 (http://www.ncbi.nlm.nih.gov/protei ...
在蛋白质组学研究中,如果使用高通量方法会得到大量蛋白质数据, 这就需要采用生物信息学的方法进行处理. 这里介绍一篇文章,希望能起到抛砖引玉的作用, 让大家讨论一下还可以用那些方法进行生物信息学处理.在这篇论文中, 应用了合并2种检索, 非标记定量, 相对量比较(normalized and non normalized),GO term 比较, 3种算法的蛋白定位预测比较, 通路分析,蛋白修饰(包括氨基酸修饰,和蛋白降解修饰)。另外在结果表格中还列出信号肽, 跨膜区,以及 ...
由于是新手,且前次在实验间隙整理,时间仓促,有诸多不足,现重新整理如下:SDS-PAGE前两条Marker样品弥撒原因SDS-PAGE电泳中溴酚兰指示带弥散的问题电泳时蛋白条带弥散蛋白质跑不到分离胶中SDS-PAGE怎么跑不出带?蛋白条带丢失的原因电泳快速脱色及染色从sds胶里面回收蛋白的几个相关问题SDS-PAGE电泳后可以回收再复性吗?用什么方法保存二维电泳SDS胶?SDS聚丙烯酰胺凝胶的干燥保存方法等电 ...
关于丁香通
公司信息
个人用户
企业机构


