


- ¥1580 - 2480
- gelatins
- jlcR12388
- 国内
- 2025年07月12日
- WB,ELISA等
- 人/动物/植物

企业认证
相关产品推荐更多 >




万千商家帮你免费找货
0 人在求购买到急需产品
- 详细信息
- 文献和实验
- 技术资料
- 供应商:
江西江蓝纯生物试剂有限公司
- 库存:
24
- 克隆性:
单克隆
- 保质期:
1年
- 抗体英文名:
KIF7
- 抗体名:
驱动蛋白家族蛋白7抗体
- 适应物种:
人/动物/植物
- 应用范围:
WB,ELISA等
- 浓度:
1mg/ml
- 保存条件:
-20 °
- 规格:
100ul/200ul
| 规格: | 100ul | 产品价格: | ¥1580.0 |
|---|---|---|---|
| 规格: | 200ul | 产品价格: | ¥2480.0 |
英文名称 : KIF7
中文名称 : 驱动蛋白家族蛋白7抗体
别 名 : EQYK340; kif 7; Kif-7; KIF7_HUMAN; kinesin family member 7; kinesin like protein KIF7; Kinesin-like protein kif7; UNQ340.
研究领域 : 细胞生物 干细胞 细胞骨架
抗体来源 : Rabbit
克隆类型 : Polyclonal
交叉反应 : Human, Mouse, Rat, Dog,
产品应用 : WB=1:500-2000 ELISA=1:500-1000 IHC-P=1:400-800 IHC-F=1:400-800 ICC=1:100-500 IF=1:100-500 (石蜡切片需做抗原修复)
not yet tested in other applications.
optimal dilutions/concentrations should be determined by the end user.
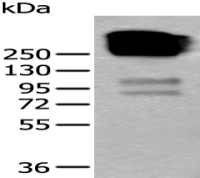
分 子 量 : 150kDa
细胞定位 : 细胞浆
性 状 : Lyophilized or Liquid
浓 度 : 1mg/ml
免 疫 原 : KLH conjugated synthetic peptide derived from human KIF7:751-850/1343
亚 型 : IgG
纯化方法 : affinity purified by Protein A
储 存 液 : 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol.
保存条件 : Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C.
PubMed : PubMed
产品介绍 : KIF7 is a 1,343 amino acid protein expressed in embryonic stem cells, melanotic melanoma and Jurkat T-cells. KIF7 is a member of the KIF27 subfamily of the kinesin-like protein family and contains one kinesin-motor domain. It is suggested that KIF7 may participate in the Hedgehog (Hh) signaling pathway by regulating the proteolysis and stability of GLI transcription factors. Hedgehog (Hh) signaling plays a critical role in embryonic development.
Function:
Acts as both a negative and positive regulator of sonic hedgehog (Shh) pathway, acting downstream of SMO. Negatively regulates the pathway by preventing inappropriate activation of the transcriptional activator GLI2 in the absence of ligand. Positively regulates the pathway by preventing the processing of the transcription factor GLI3 into its repressor form. Required for efficient localization of GLI3 to cilia in response to Shh. May also act as a ciliary motor.
Subunit:
Interacts with GLI1, GLI2, GLI3, SMO and SUFU. Interacts with NPHP1.
Subcellular Location:
Cell projection; cilium. SMO is required for its accumulation within cilia. Moves from the cilia base to the cilia tip in response to activation of the Shh pathway.
Tissue Specificity:
Embryonic stem cells, melanotic melanoma and Jurkat T-cells.
DISEASE:
Note=Ciliary dysfunction leads to a broad spectrum of disorders, collectively termed ciliopathies. The ciliopathy range of diseases includes Meckel-Gruber syndrome, Bardet-Biedl syndrome, Joubert syndrome, and hydrolethalus syndrome among others. Single-locus allelism is insufficient to explain the variable penetrance and expressivity of such disorders, leading to the suggestion that variations across multiple sites of the ciliary proteome influence the clinical outcome. Primary ciliopathy loci can be modulated by pathogenic lesions in other ciliary genes to either exacerbate overall severity or induce specific endophenotypes. KIF7 may be causally associated with diverse ciliopathies, and also acts as a modifier gene across the ciliopathy spectrum.
Defects in KIF7 may be a cause of Bardet-Biedl syndrome (BBS) [MIM:209900]. A syndrome characterized by usually severe pigmentary retinopathy, early-onset obesity, polydactyly, hypogenitalism, renal malformation and mental retardation. Secondary features include diabetes mellitus, hypertension and congenital heart disease. Bardet-Biedl syndrome inheritance is autosomal recessive, but three mutated alleles (two at one locus, and a third at a second locus) may be required for clinical manifestation of some forms of the disease. Note=Heterozygous missense mutations in KIF7 may genetically interact with other BBS genes and contribute to disease manifestation and severity.
Defects in KIF7 are the cause of hydrolethalus syndrome type 2 (HLS2) [MIM:614120]. HLS2 is an embryonic lethal disorder characterized by hydrocephaly or anencephaly, postaxial polydactyly of the upper limbs, and pre- or postaxial polydactyly of the lower limbs. Duplication of the hallux is a common finding.
Defects in KIF7 are the cause of acrocallosal syndrome (ACLS) [MIM:200990]. ACLS is a syndrome that is characterized by postaxial polydactyly, hallux duplication, macrocephaly and absence of the corpus callosum, usually with severe developmental delay.
Defects in KIF7 are the cause of Joubert syndrome type 12 (JBTS12) [MIM:200990]. JBTS12 is a disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy and renal disease.
Defects in KIF7 may be a cause of Pallister-Hall syndrome (PHS) [MIM:146510]. An autosomal dominant disorder characterized by a wide range of clinical manifestations. Clinical features include hypothalamic hamartoma, pituitary dysfunction, central or postaxial polydactyly, and syndactyly. Malformations are frequent in the viscera, e.g. anal atresia, bifid uvula, congenital heart malformations, pulmonary or renal dysplasia.
Similarity:
Belongs to the kinesin-like protein family. KIF27 subfamily.
Contains 1 kinesin-motor domain.
SWISS:
Q2M1P5
Gene ID:
374654
Important Note:
This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications.
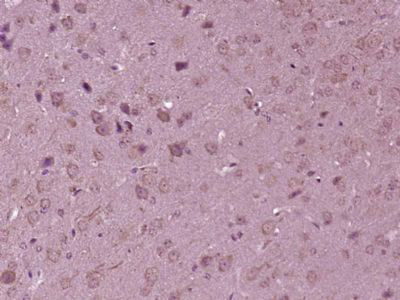
产品图片
风险提示:丁香通仅作为第三方平台,为商家信息发布提供平台空间。用户咨询产品时请注意保护个人信息及财产安全,合理判断,谨慎选购商品,商家和用户对交易行为负责。对于医疗器械类产品,请先查证核实企业经营资质和医疗器械产品注册证情况。
 文献和实验
文献和实验驱动蛋白是1985年从鱿鱼的轴质(axonplasm)中分离的一种发动机蛋白。驱动蛋白是一个大的复合蛋白,由几个不同的结构域组成, 包括两条重链和一条轻链, 总分子量为380kDa。它有一对球形的头,是产生动力的“电机”; 还有一个扇形的尾,是货物结合部位。 体外实验证明驱动蛋白的运输具有方向性,从微管的(-)端移向微管的( )端,是正端走向的微管发动机(plus end-directed microtublar motor)。
生理状态或病理状态下蛋白水平的量变,微型化,集成化,高通量化的抗体芯片就是一个非常好的研究工具,它也是蛋白芯片中发展最快的芯片,而且在技术上已经日益成熟。这些抗体芯片有的已经在向临床应用上发展,比如肿瘤标志物抗体芯片等,还有很多已经用在研究的各个领域里。第一张商品化的抗体芯片是由美国BD Clontech公司推出的。这是一张用于研究的抗体芯片,芯片上排列了378种已知蛋白的单抗(Ab Microarray 380,目录号 K1847-1),这些单抗对应的蛋白都是细胞结构和功能上十分重要的蛋白,涉及信号
人 抗麦胶蛋白抗体 ( AGA) 酶联免疫分析 试剂盒使用说明书 本试剂仅供研究使用 目的:本试剂盒用于测定人血清,血浆及相关液体样本中 抗麦胶蛋白抗体( AGA) 的 含量。 实验原理: 本试剂盒应用双抗原夹心法测定标本中人 抗麦胶蛋白抗体( AGA) 的 水平。用纯化的 抗原 包被微孔板,制成固相抗原,往包被抗原的微孔中依次加入 抗麦胶蛋白抗体( AGA) ,再与 HRP 标记的 抗原 结合
 技术资料
技术资料暂无技术资料 索取技术资料

