大家都在搜

手机验证

询价列表

暂时没有已询价产品

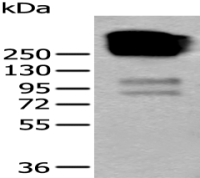
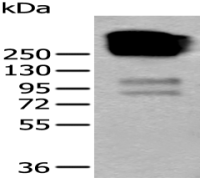
间隙连接蛋白26/GJB2抗体

企业认证
江西江蓝纯生物试剂有限公司
159
单克隆
1年
Connexin 26
间隙连接蛋白26/GJB2抗体
人/动物/植物
WB,ELISA等
1mg/ml
-20 °
100ul/200ul
| 规格: | 100ul | 产品价格: | ¥1580.0 |
|---|---|---|---|
| 规格: | 200ul | 产品价格: | ¥2480.0 |

风险提示:丁香通仅作为第三方平台,为商家信息发布提供平台空间。用户咨询产品时请注意保护个人信息及财产安全,合理判断,谨慎选购商品,商家和用户对交易行为负责。对于医疗器械类产品,请先查证核实企业经营资质和医疗器械产品注册证情况。
 技术资料
技术资料
需要更多技术资料 索取更多技术资料
间隙连接蛋白26GJB2抗体.doc 附 (下载 0 次)