大家都在搜

手机验证

询价列表

暂时没有已询价产品
Illumina全转录组测序
Illumina全转录组测序
简介
全转录组测序采用长读长对读测序和更高的测序深度,可全面快速的获取某一条件下组织或细胞绝大部分转录本的定性和定量情况。与表达谱研究相比,全转录组测序除了可对转录本进行更加准确的定量外,还可进行更为复杂的序列分析,如新转录本和罕见转录本发现、cSNP和InDel鉴定、可变剪切鉴定、lncRNA和UTRs鉴定等,获得更为精确完整的基因功能图谱和突破性的发现。
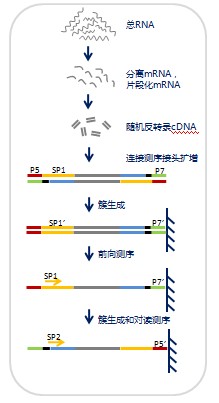
实验流程
|
|
1.样品提取总RNA后,过柱纯化,保留200nt以上的RNA。 2.富集mRNA,并用二价阳离子高温加热的方法将mRNA片断化。 3.以mRNA短片段为模板,用六碱基随机引物反转录合成双链cDNA。 4.经纯化、末端修复、加碱基A、加测序接头的步骤,根据测序长度和是否做对读测序,确定琼脂糖凝胶电泳回收片段的大小,并进行PCR扩增,完成测序文库的制备。 5.构建好的文库经cBot扩增,用Illumina Hiseq 2000进行单端或对读测序。 |
技术优势
精确性高:精确到单核苷酸水平,可区分高度同源的RNA分子,鉴定剪切变异体和RNA多态性。
高度开放性:无需预先了解序列信息,可研究任一物种已知和未知的转录本序列及定量信息。
灵敏度高:可检测低丰度转录本。
检测范围广:可在超过6个数量级范围内实现准确的序列检测和定量分析。
灵活性高:可根据实验目的灵活选择合适的测序reads深度和reads读长,以达到对不同丰度转录本及序列变异的检测要求。
高质量的数据:采取长读长对读测序及更高的测序深度,提高了对重复序列和同源序列的辨识能力,有效提高可绘制转录组的比例和转录组装的可信度,获得更多长无误的重叠群contigs。
均匀的覆盖度:采用随机降解转录本和随机反转录的策略制备测序文库,有利于获得更为均匀的读取分布,提高了测序效率。
获取更丰富的转录信息:更高的测序深度和更均匀的覆盖度,帮助研究者发现和区分低丰度的转录本、高度相似的转录本、可变剪切及单个核苷酸多态性。
样品要求
样品类型:细胞、新鲜组织或RNA样品。
样品量:细胞样品请提供至少1×107个细胞,组织样品请提供至少300mg的组织块或切片,RNA样品请提供30 μg以上的总RNA。
样品质量:RNA无明显降解,提取的总RNA OD260/280值在1.8~2.2之间,浓度≥500 ng/μl,28S:18S≥1.5,RIN≥8。
样品保存:细胞样品或新鲜组织块(切成~50mg的小块)可用TRIZOL或RNA保护剂处理或液氮冻存后,-80℃保存。RNA样品可溶于乙醇或RNA-free的超纯水中,-80℃保存。样品保存期间避免反复冻融。
样品运输:样品置于1.5 ml管中,封口膜封好,干冰运输。
数据分析
1.无参考基因组的转录组分析(De novo Transcriptome Analysis)
Contig和Unigene统计分析
Unigene功能注释((NT, NR, Swissprot, Kegg, COG, Interpro&GO)
Unigene可变剪切分析
Unigene表达差异分析(不少于两个样品)
2.有参考基因组的转录组分析(Transcriptome analysis with reference genome)
数据产出统计及质量评估
Reads与参考基因组序列或转录组序列比对
Reads在基因组上的分布统计
反义转录本注释
转录本定量
基因功能注释
基因GO和pathway显著性富集分析
基因表达差异分析(不少于两个样品)及聚类分析
基因结构及变异分析:可变剪切、基因融合及cSNP鉴定等
预测新基因
参考文献
1.Mortazavi A, Williams B, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods, 2008; 5: 621-628.
2.Maher CA, Kumar-Sinha C, Cao X, et al. Transcriptome Sequencing to Detect Gene Fusions in Cancer.Nature, 2009; 458: 97-101.
3.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet, 2009; 10: 57-63.
4.Zhang G, Guo G, Hu X, et al. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res, 2010; 20: 646-654.
5.Cirulli ET, Singh A, Shianna1 KV.Screening the human exome: a comparison of whole genome and whole transcriptome sequencing. Genome Biol, 2010; 11: R57.
6.Palanisamy N, Ateeq B, Kalyana-Sundaram S, et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nature Med, 2010; 16: 793-798.
7.Illumina white paper for sequencing: RNA-Seq data comparison with gene expression microarrays.
风险提示:丁香通仅作为第三方平台,为商家信息发布提供平台空间。用户咨询产品时请注意保护个人信息及财产安全,合理判断,谨慎选购商品,商家和用户对交易行为负责。对于医疗器械类产品,请先查证核实企业经营资质和医疗器械产品注册证情况。
 询价记录
询价记录