大家都在搜

手机验证

询价列表

暂时没有已询价产品
Collagen alpha-
1(I) chain I型胶原蛋白
企业认证
上海联迈生物工程有限公司
大量
LM-20124R
多克隆
Rabbit
1年
Collagen alpha-1(I) chain
I型胶原蛋白
Rabbit
Human, Mouse, Rat,
KLH conjugated synthetic peptide derived from human Collagen alpha-1(I) chain:1051-1218
IgG
Lyophilized or Liquid
ELISA=1:500-1000 IHC-P=1:400-800 IHC-F=1:400-800 ICC=1:100-500 IF=1:100-500 (石蜡切片需做抗原修复)
1mg/ml
Store at -20 °C
100ul 200ul
| 英文名称 | Collagen alpha-1(I) chain |
| 中文名称 | I型胶原蛋白 |
| 别 名 | Collagen type I; Alpha 1 type I collagen; COL1A1; Collagen I alpha 1 polypeptide; Collagen Of Skin Tendon And Bone; Collagen Type 1; Collagen type I alpha 1; OI4; Osteogenesis Imperfecta Type IV; Pro alpha 1(I) collagen; Type I procollagen; CO1A1_HUMAN; Chondrocalcin; collagen alpha-1(I) chain preproprotein. 胶原蛋白1; 1型胶原蛋白; I型胶原 |
| 规格价格 | 100ul/1380元 购买 200ul/2200元 购买 大包装/询价 |
| 说 明 书 | 100ul 200ul |
| 研究领域 | 细胞生物 免疫学 |
| 抗体来源 | Rabbit |
| 克隆类型 | Polyclonal |
| 交叉反应 | Human, Mouse, Rat, |
| 产品应用 | ELISA=1:500-1000 IHC-P=1:400-800 IHC-F=1:400-800 ICC=1:100-500 IF=1:100-500 (石蜡切片需做抗原修复) not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. |
| 分 子 量 | 116kDa |
| 细胞定位 | 细胞外基质 分泌型蛋白 |
| 性 状 | Lyophilized or Liquid |
| 浓 度 | 1mg/ml |
| 免 疫 原 | KLH conjugated synthetic peptide derived from human Collagen alpha-1(I) chain:1051-1218 |
| 亚 型 | IgG |
| 纯化方法 | affinity purified by Protein A |
| 储 存 液 | Preservative: 15mM Sodium Azide, Constituents: 1% BSA, 0.01M PBS, pH 7.4 |
| 保存条件 | Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C. |
| PubMed | PubMed |
| 产品介绍 | background: This gene encodes the pro-alpha1 chains of type I collagen whose triple helix comprises two alpha1 chains and one alpha2 chain. Type I is a fibril-forming collagen found in most connective tissues and is abundant in bone, cornea, dermis and tendon. Mutations in this gene are associated with osteogenesis imperfecta types I-IV, Ehlers-Danlos syndrome type VIIA, Ehlers-Danlos syndrome Classical type, Caffey Disease and idiopathic osteoporosis. Reciprocal translocations between chromosomes 17 and 22, where this gene and the gene for platelet-derived growth factor beta are located, are associated with a particular type of skin tumor called dermatofibrosarcoma protuberans, resulting from unregulated expression of the growth factor. Two transcripts, resulting from the use of alternate polyadenylation signals, have been identified for this gene. [provided by R. Dalgleish, Feb 2008]. Function: Type I collagen is a member of group I collagen (fibrillar forming collagen). Subunit: Trimers of one alpha 2(I) and two alpha 1(I) chains. Interacts with MRC2. Interacts with TRAM2. Subcellular Location: Secreted, extracellular space, extracellular matrix. Tissue Specificity: Forms the fibrils of tendon, ligaments and bones. In bones the fibrils are mineralized with calcium hydroxyapatite. Post-translational modifications: Proline residues at the third position of the tripeptide repeating unit (G-X-P) are hydroxylated in some or all of the chains. Proline residues at the second position of the tripeptide repeating unit (G-P-X) are hydroxylated in some of the chains. O-linked glycan consists of a Glc-Gal disaccharide bound to the oxygen atom of a post-translationally added hydroxyl group. DISEASE: Defects in COL1A1 are the cause of Caffey disease (CAFFD) [MIM:114000]; also known as infantile cortical hyperostosis. Caffey disease is characterized by an infantile episode of massive subperiosteal new bone formation that typically involves the diaphyses of the long bones, mandible, and clavicles. The involved bones may also appear inflamed, with painful swelling and systemic fever often accompanying the illness. The bone changes usually begin before 5 months of age and resolve before 2 years of age. Defects in COL1A1 are a cause of Ehlers-Danlos syndrome type 1 (EDS1) [MIM:130000]; also known as Ehlers-Danlos syndrome gravis. EDS is a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDS1 is the severe form of classic Ehlers-Danlos syndrome. Defects in COL1A1 are the cause of Ehlers-Danlos syndrome type 7A (EDS7A) [MIM:130060]; also known as autosomal dominant Ehlers-Danlos syndrome type VII. EDS is a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDS7A is marked by bilateral congenital hip dislocation, hyperlaxity of the joints, and recurrent partial dislocations. Defects in COL1A1 are a cause of osteogenesis imperfecta type 1 (OI1) [MIM:166200]. A dominantly inherited connective tissue disorder characterized by bone fragility and blue sclerae. Osteogenesis imperfecta type 1 is non-deforming with normal height or mild short stature, and no dentinogenesis imperfecta. Defects in COL1A1 are a cause of osteogenesis imperfecta type 2 (OI2) [MIM:166210]; also known as osteogenesis imperfecta congenita. A connective tissue disorder characterized by bone fragility, with many perinatal fractures, severe bowing of long bones, undermineralization, and death in the perinatal period due to respiratory insufficiency. Defects in COL1A1 are a cause of osteogenesis imperfecta type 3 (OI3) [MIM:259420]. A connective tissue disorder characterized by progressively deforming bones, very short stature, a triangular face, severe scoliosis, grayish sclera, and dentinogenesis imperfecta. Defects in COL1A1 are a cause of osteogenesis imperfecta type 4 (OI4) [MIM:166220]; also known as osteogenesis imperfecta with normal sclerae. A connective tissue disorder characterized by moderately short stature, mild to moderate scoliosis, grayish or white sclera and dentinogenesis imperfecta. Genetic variations in COL1A1 are a cause of susceptibility to osteoporosis (OSTEOP) [MIM:166710]; also known as involutional or senile osteoporosis or postmenopausal osteoporosis. Osteoporosis is characterized by reduced bone mass, disruption of bone microarchitecture without alteration in the composition of bone. Osteoporotic bones are more at risk of fracture. Note=A chromosomal aberration involving COL1A1 is found in dermatofibrosarcoma protuberans. Translocation t(17;22)(q22;q13) with PDGF. Similarity: Belongs to the fibrillar collagen family. Contains 1 fibrillar collagen NC1 domain. Contains 1 VWFC domain. SWISS: P02452 Gene ID: 1277 Database links: Entrez Gene: 1277 Human Entrez Gene: 1278 Human Entrez Gene: 12842 Mouse Entrez Gene: 12843 Mouse Entrez Gene: 29393 Rat Entrez Gene: 84352 Rat Omim: 120150 Human Omim: 120160 Human SwissProt: P02452 Human SwissProt: P08123 Human SwissProt: P11087 Mouse SwissProt: Q01149 Mouse SwissProt: P02454 Rat SwissProt: P02466 Rat Unigene: 172928 Human Unigene: 489142 Human Unigene: 681002 Human Unigene: 277735 Mouse Unigene: 277792 Mouse Unigene: 458212 Mouse Unigene: 107239 Rat Unigene: 2953 Rat Important Note: This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. I型胶原是软骨基质中的一种结构蛋白,主要用于Ⅰ型胶原分布及变态反应方面的研究。 取消WB;姬,2012.12.27; |
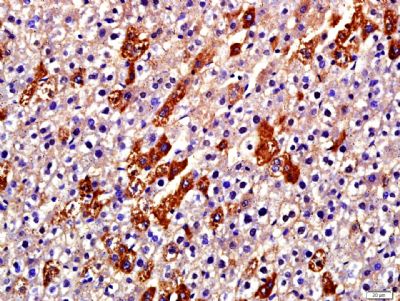
| 产品图片 |  Paraformaldehyde-fixed, paraffin embedded (Rat liver); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (Collagen alpha-1(I) chain) Polyclonal Antibody, Unconjugated (bs-20124R) at 1:500 overnight at 4°C, followed by a conjugated secondary (sp-0023) for 20 minutes and DAB staining. |
风险提示:丁香通仅作为第三方平台,为商家信息发布提供平台空间。用户咨询产品时请注意保护个人信息及财产安全,合理判断,谨慎选购商品,商家和用户对交易行为负责。对于医疗器械类产品,请先查证核实企业经营资质和医疗器械产品注册证情况。