
大家都在搜

手机验证

询价列表

暂时没有已询价产品

BioXCell代理|BioX
Cell现货库存中心
企业认证
Bioxcell抗体
Bioxcell antibody
多样
多样
广泛
广泛
欣博盛生物
大量现货
体内研究
BE或BP开头
广泛
一年
小鼠/大鼠
广泛
单克隆
产品说明
产品说明
产品说明
产品说明
1/5/25/50/100mg
BioXCell代理,BioXCell现货库存中心 --欣博盛生物 BioXCell的中和/阻断抗体受到国内外研究者的普遍热爱和青睐,在肿瘤、癌症等方面的研究中广受好评。BioXCell公司位于美国新罕布什尔州,具有25年以上单克隆抗体和重组蛋白的生产及定制经验,可提供高纯度、低内毒素、无防腐剂、适用于体内临床前研究的单克隆抗体。
欣博盛生物是Bioxcell一级代理商,Bioxcell国内唯一库存中心。如需购买Bioxcell公司产品,请联系我们。
 |
高纯度、低内毒素,无防腐剂,适用于体内临床前研究 |
| 超过二十五年单克隆抗体和重组蛋白生产及定制经验 | |
| 性价比高,可提供100mg甚至50g的大包装 | |
| 超过15000篇高质量期刊文献引用 |
肿瘤癌症免疫治疗应用
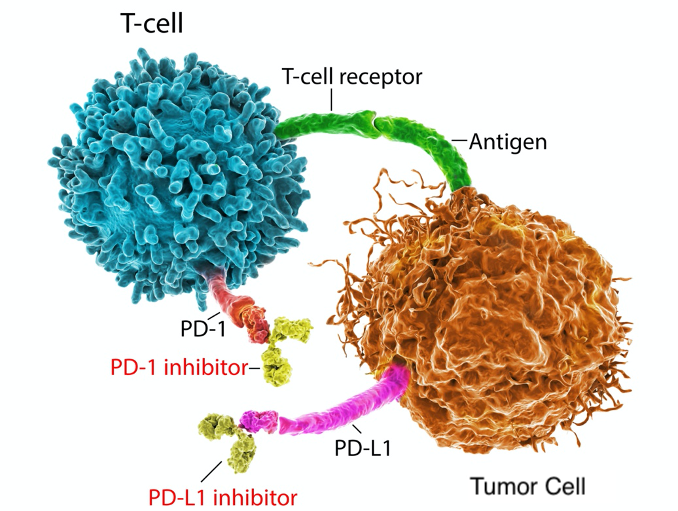
●抑制免疫检查点和其他免疫调节来治疗恶性肿瘤是现在利用免疫系统杀死肿瘤细胞的有力途径。BioXCell提供三种不同克隆号的anti mouse PD-1抗体:RMP1-14、29F.1A12和J43。三种抗体都通过相同的机制发挥作用—它们结合PD-1,并在空间上阻断PD-1与PD-1配体的结合,从而阻断PD-1信号传导。这三种克隆号的抗体都非常适合在小鼠体内模型中阻断PD-1信号传导,并有大量文献支持这一应用。这些抗体之间的差异在于文献报道中的其他应用、Isotype和来源。
●通过用阻断PD-L1和它的受体PD-1之间的相互作用的抗体治疗,肿瘤生长可以暂时被抑制。
●抗PD-1联合抗CTLA-4抗体介导的免疫治疗对黑色素瘤、肾细胞癌和非小细胞肺癌具有显著疗效。
●通过靶向调节免疫反应的途径,如RANK途径,可以将“cold”肿瘤转化为“hot”肿瘤。使用Bio X Cell’s Anti-mouse RANKL (clone IK22/5) antibody,研究人员证明了对RANK通路的抑制将“cold”乳腺肿瘤转化为“hot”肿瘤,变为“hot”的肿瘤可能受益于免疫疗法。
InVivoMab vs. InVivoPlus 如何选择?
| InVivoMab |
InVivoPlus |
|
| 纯度 |
> 95% |
> 95% |
| 蛋白完整性 |
√ (verified via SDS-PAGE) |
ß(verified via SDS-PAGE) |
| 内毒素浓度 |
< 2EU/mg |
< 1EU/mg |
| 不含叠氮化物和载体蛋白 |
√ |
√ |
| 是否适用于体内研究 |
√ |
√ |
| 是否提供大包装 |
√ |
√ |
| 是否经WB, FC或ELISA验证 |
√ |
|
| 经验证蛋白聚集≤ 5% |
√ |
|
| 是否经过鼠科病原体检测 |
√ |
|
| 产品货号 |
BE-开头 |
BP-开头 |
这些指标的抗体最畅销!
肿瘤研究抗体
| 抗原 |
应用 |
InVivoMab 目录号 |
InVivoPlus 目录号 |
| PD-1 (CD279) |
in vivo blocking of PD-1/PD-L signaling, WB |
BE0146 |
BP0146
|
| in vivo blocking of PD-1/PD-L signaling, in vitro PD-1 neutralization |
BE0033-2 |
BP0033-2
|
|
| PD-L1(B7-H1)
|
in vivo PD-L1 blockade, IF, IHC-F, FC, WB
|
BE0101
|
BP0101
|
| CTLA-4 (CD152)
|
in vivo CTLA-4 neutralization, WB
|
BE0164 |
BP0164
|
| in vivo and in vitro CTLA-4 neutralization |
BE0131 |
BP0131 |
|
| CD4 |
in vivo CD4+ T cell depletion, FC
|
BE0003-1 |
BP0003-1
|
| Ly6G |
in vivo neutrophil depletion, in vivo MDSC depletion, IF, IHC-P, IHC-F, FC |
BE0075-1 |
BP0075-1 |
| in vivo depletion of Gr-1+ myeloid cells, FC |
BE0075 |
BP0075 |
|
| CD8α
|
in vivo CD8+T cell depletion
|
BE0061 BE0117 |
BP0061 BP0117 |
| in vivo CD8+ T cell depletion, IF, FC
|
BE0004-1 |
BP0004-1
|
|
| OX40 (CD134)
|
in vivo and in vitro OX40 activation
|
BE0031 |
|
| CSF1R (CD115)
|
in vivo macrophage/monocyte depletion, in vitro CSF-R1 neutralization, FC
|
BE0213 |
|
| CD40 |
in vivoCD40 activation, in vitro B cell stimulation, in vitro DC stimulation
|
BE0016-2 |
BP0016-2
|
| IFNγ
|
in vivo and in vitro IFNγ neutralization, ELISPOT, FC |
BE0055 |
BP0055 |
| GITR
|
in vivo GITR stimulation
|
BE0063 |
|
| CD154 (CD40L)
|
in vivo and in vitro blocking of CD40/CD40L signaling
|
BE0017-1 |
BP0017-1
|
| NK1.1
|
in vivo NK cell depletion, FC
|
BE0036 |
BP0036
|
| TGF-β
|
in vivo TGFβ neutralization, in vitro TGFβ neutralization
|
BE0057 |
BP0057
|
畅销同型对照抗体
| 同型对照 |
InVivoMab 目录号 |
InVivoPlus 目录号 |
| Rat IgG2a Isotype control, FC |
BE0089 |
BP0089 |
| Rat IgG2b Isotype control |
BE0090 |
BP0090 |
| Mouse IgG1 Isotype control |
BE0083 |
BP0083 |
| Rat IgG1 Isotype control |
BE0088 |
BP0088 |
| Mouse IgG2a Isotype control |
BE0085 |
BP0085 |
| Mouse IgG2b Isotype control |
BE0086 |
BP0086 |
| Polyclonal Armenian Hamster IgG |
BE0091 |
BP0091 |
免疫细胞特异性标志物抗体(Immune Cell Specific Depletion Antibodies)
Bioxcell还可提供各种标签抗体
| 产品名称 |
产品编号 |
| ReadyTag anti-c-myc |
RT0263 |
| ReadyTag anti-GST |
RT0264 |
| ReadyTag anti-GFP |
RT0265 |
| ReadyTag anti-6-His |
RT0266 |
| ReadyTag anti-OVA |
RT0267 |
| ReadyTag anti-HA |
RT0268 |
| ReadyTag anti-DDDDK |
RT0269 |
Bioxcell产品清单(部分):
如需获取BIoxcell产品折扣报价,请联系欣博盛生物!
| 品牌 | 产品编号 | 产品名称 | 规格 |
| BioXcell | BE0000-1MG | InVivoMAb anti-mouse CD1d (CD1.1) | 1 mg |
| BioXcell | BE0000-5MG | InVivoMAb anti-mouse CD1d (CD1.1) | 5 mg |
| BioXcell | BE0000-25MG | InVivoMAb anti-mouse CD1d (CD1.1) | 25 mg |
| BioXcell | BE0000-50MG | InVivoMAb anti-mouse CD1d (CD1.1) | 50 mg |
| BioXcell | BE0000-100MG | InVivoMAb anti-mouse CD1d (CD1.1) | 100 mg |
| BioXcell | BE0001-1-1MG | InVivoMAb anti-mouse CD3ε | 1 mg |
| BioXcell | BE0001-1-5MG | InVivoMAb anti-mouse CD3ε | 5 mg |
| BioXcell | BE0001-1-25MG | InVivoMAb anti-mouse CD3ε | 25 mg |
| BioXcell | BE0001-1-50MG | InVivoMAb anti-mouse CD3ε | 50 mg |
| BioXcell | BE0001-1-100MG | InVivoMAb anti-mouse CD3ε | 100 mg |
| BioXcell | BE0001-1FAB-10MG | InVivoMAb anti-mouse CD3ε F(ab')2 fragment | 10 mg |
| BioXcell | BE0001-1FAB-5MG | InVivoMAb anti-mouse CD3ε F(ab')2 fragment | 5 mg |
| BioXcell | BE0001-1FAB-25MG | InVivoMAb anti-mouse CD3ε F(ab')2 fragment | 25 mg |
| BioXcell | BE0001-1FAB-50MG | InVivoMAb anti-mouse CD3ε F(ab')2 fragment | 50 mg |
| BioXcell | BE0001-2-1MG | InVivoMAb anti-human CD3 | 1 mg |
| BioXcell | BE0001-2-5MG | InVivoMAb anti-human CD3 | 5 mg |
| BioXcell | BE0001-2-25MG | InVivoMAb anti-human CD3 | 25 mg |
| BioXcell | BE0001-2-50MG | InVivoMAb anti-human CD3 | 50 mg |
| BioXcell | BE0001-2-100MG | InVivoMAb anti-human CD3 | 100 mg |
| BioXcell | BE0002-1MG | InVivoMAb anti-mouse CD3 | 1 mg |
| BioXcell | BE0002-5MG | InVivoMAb anti-mouse CD3 | 5 mg |
| BioXcell | BE0002-25MG | InVivoMAb anti-mouse CD3 | 25 mg |
| BioXcell | BE0002-50MG | InVivoMAb anti-mouse CD3 | 50 mg |
| BioXcell | BE0002-100MG | InVivoMAb anti-mouse CD3 | 100 mg |
| BioXcell | BE0003-1-1MG | InVivoMAb anti-mouse CD4 | 1 mg |
| BioXcell | BE0003-1-5MG | InVivoMAb anti-mouse CD4 | 5 mg |
| BioXcell | BE0003-1-25MG | InVivoMAb anti-mouse CD4 | 25 mg |
| BioXcell | BE0003-1-50MG | InVivoMAb anti-mouse CD4 | 50 mg |
| BioXcell | BE0003-1-100MG | InVivoMAb anti-mouse CD4 | 100 mg |
| BioXcell | BE0003-2-1MG | InVivoMAb anti-human CD4 | 1 mg |
| BioXcell | BE0003-2-5MG | InVivoMAb anti-human CD4 | 5 mg |
| BioXcell | BE0003-2-25MG | InVivoMAb anti-human CD4 | 25 mg |
| BioXcell | BE0003-2-50MG | InVivoMAb anti-human CD4 | 50 mg |
| BioXcell | BE0003-2-100MG | InVivoMAb anti-human CD4 | 100 mg |
| BioXcell | BE0003-3-1MG | InVivoMAb anti-mouse CD4 | 1 mg |
| BioXcell | BE0003-3-5MG | InVivoMAb anti-mouse CD4 | 5 mg |
| BioXcell | BE0003-3-25MG | InVivoMAb anti-mouse CD4 | 25 mg |
| BioXcell | BE0003-3-50MG | InVivoMAb anti-mouse CD4 | 50 mg |
| BioXcell | BE0003-3-100MG | InVivoMAb anti-mouse CD4 | 100 mg |
| BioXcell | BE0004-1-1MG | InVivoMAb anti-mouse CD8α | 1 mg |
| BioXcell | BE0004-1-5MG | InVivoMAb anti-mouse CD8α | 5 mg |
| BioXcell | BE0004-1-25MG | InVivoMAb anti-mouse CD8α | 25 mg |
| BioXcell | BE0004-1-50MG | InVivoMAb anti-mouse CD8α | 50 mg |
| BioXcell | BE0004-1-100MG | InVivoMAb anti-mouse CD8α | 100 mg |
| BioXcell | BE0004-2-1MG | InVivoMAb anti-human CD8α | 1 mg |
| BioXcell | BE0004-2-5MG | InVivoMAb anti-human CD8α | 5 mg |
| BioXcell | BE0004-2-25MG | InVivoMAb anti-human CD8α | 25 mg |
| BioXcell | BE0004-2-50MG | InVivoMAb anti-human CD8α | 50 mg |
| BioXcell | BE0004-2-100MG | InVivoMAb anti-human CD8α | 100 mg |
| BioXcell | BE0005-1MG | InVivoMAb anti-human LFA-1α (CD11a) | 1 mg |
| BioXcell | BE0005-5MG | InVivoMAb anti-human LFA-1α (CD11a) | 5 mg |
| BioXcell | BE0005-25MG | InVivoMAb anti-human LFA-1α (CD11a) | 25 mg |
| BioXcell | BE0005-50MG | InVivoMAb anti-human LFA-1α (CD11a) | 50 mg |
| BioXcell | BE0005-100MG | InVivoMAb anti-human LFA-1α (CD11a) | 100 mg |
| BioXcell | BE0005-1-1MG | InVivoMAb anti-mouse LFA-1α (CD11a) | 1 mg |
| BioXcell | BE0005-1-5MG | InVivoMAb anti-mouse LFA-1α (CD11a) | 5 mg |
| BioXcell | BE0005-1-25MG | InVivoMAb anti-mouse LFA-1α (CD11a) | 25 mg |
| BioXcell | BE0005-1-50MG | InVivoMAb anti-mouse LFA-1α (CD11a) | 50 mg |
| BioXcell | BE0005-1-100MG | InVivoMAb anti-mouse LFA-1α (CD11a) | 100 mg |
| BioXcell | BE0006-1MG | InVivoMAb anti-mouse LFA-1α (CD11a) | 1 mg |
| BioXcell | BE0006-5MG | InVivoMAb anti-mouse LFA-1α (CD11a) | 5 mg |
| BioXcell | BE0006-25MG | InVivoMAb anti-mouse LFA-1α (CD11a) | 25 mg |
| BioXcell | BE0006-50MG | InVivoMAb anti-mouse LFA-1α (CD11a) | 50 mg |
| BioXcell | BE0006-100MG | InVivoMAb anti-mouse LFA-1α (CD11a) | 100 mg |
| BioXcell | BE0007-1MG | InVivoMAb anti-mouse/human CD11b | 1 mg |
| BioXcell | BE0007-5MG | InVivoMAb anti-mouse/human CD11b | 5 mg |
| BioXcell | BE0007-25MG | InVivoMAb anti-mouse/human CD11b | 25 mg |
| BioXcell | BE0007-50MG | InVivoMAb anti-mouse/human CD11b | 50 mg |
| BioXcell | BE0007-100MG | InVivoMAb anti-mouse/human CD11b | 100 mg |
| BioXcell | BE0009-1MG | InVivoMAb anti-mouse CD18 | 1 mg |
| BioXcell | BE0009-5MG | InVivoMAb anti-mouse CD18 | 5 mg |
| BioXcell | BE0009-25MG | InVivoMAb anti-mouse CD18 | 25 mg |
| BioXcell | BE0009-50MG | InVivoMAb anti-mouse CD18 | 50 mg |
| BioXcell | BE0009-100MG | InVivoMAb anti-mouse CD18 | 100 mg |
| BioXcell | BE0011-1MG | InVivoMAb anti-mouse CD22 | 1 mg |
| BioXcell | BE0011-5MG | InVivoMAb anti-mouse CD22 | 5 mg |
| BioXcell | BE0011-25MG | InVivoMAb anti-mouse CD22 | 25 mg |
| BioXcell | BE0011-50MG | InVivoMAb anti-mouse CD22 | 50 mg |
| BioXcell | BE0011-100MG | InVivoMAb anti-mouse CD22 | 100 mg |
| BioXcell | BE0012-1MG | InVivoMAb anti-mouse CD25 (IL-2Rα) | 1 mg |
| BioXcell | BE0012-5MG | InVivoMAb anti-mouse CD25 (IL-2Rα) | 5 mg |
| BioXcell | BE0012-25MG | InVivoMAb anti-mouse CD25 (IL-2Rα) | 25 mg |
| BioXcell | BE0012-50MG | InVivoMAb anti-mouse CD25 (IL-2Rα) | 50 mg |
| BioXcell | BE0012-100MG | InVivoMAb anti-mouse CD25 (IL-2Rα) | 100 mg |
| BioXcell | BE0014-1MG | InVivoMAb anti-human CD25 (IL-2Rα) | 1 mg |
| BioXcell | BE0014-5MG | InVivoMAb anti-human CD25 (IL-2Rα) | 5 mg |
| BioXcell | BE0014-25MG | InVivoMAb anti-human CD25 (IL-2Rα) | 25 mg |
| BioXcell | BE0014-50MG | InVivoMAb anti-human CD25 (IL-2Rα) | 50 mg |
| BioXcell | BE0014-100MG | InVivoMAb anti-human CD25 (IL-2Rα) | 100 mg |
| BioXcell | BE0015-1-1MG | InVivoMAb anti-mouse CD28 | 1 mg |
| BioXcell | BE0015-1-5MG | InVivoMAb anti-mouse CD28 | 5 mg |
| BioXcell | BE0015-1-25MG | InVivoMAb anti-mouse CD28 | 25 mg |
| BioXcell | BE0015-1-50MG | InVivoMAb anti-mouse CD28 | 50 mg |
| BioXcell | BE0015-1-100MG | InVivoMAb anti-mouse CD28 | 100 mg |
| BioXcell | BE0015-5-1MG | InVivoMAb anti-mouse CD28 | 1 mg |
| BioXcell | BE0015-5-5MG | InVivoMAb anti-mouse CD28 | 5 mg |
| BioXcell | BE0015-5-25MG | InVivoMAb anti-mouse CD28 | 25 mg |
| BioXcell | BE0015-5-50MG | InVivoMAb anti-mouse CD28 | 50 mg |
| BioXcell | BE0015-5-100MG | InVivoMAb anti-mouse CD28 | 100 mg |
| BioXcell | BE0016-2-1MG | InVivoMAb anti-mouse CD40 | 1 mg |
| BioXcell | BE0016-2-5MG | InVivoMAb anti-mouse CD40 | 5 mg |
| BioXcell | BE0016-2-25MG | InVivoMAb anti-mouse CD40 | 25 mg |
| BioXcell | BE0016-2-50MG | InVivoMAb anti-mouse CD40 | 50 mg |
| BioXcell | BE0016-2-100MG | InVivoMAb anti-mouse CD40 | 100 mg |
| BioXcell | BE0017-1-1MG | InVivoMAb anti-mouse CD40L (CD154) | 1 mg |
| BioXcell | BE0017-1-5MG | InVivoMAb anti-mouse CD40L (CD154) | 5 mg |
| BioXcell | BE0017-1-25MG | InVivoMAb anti-mouse CD40L (CD154) | 25 mg |
| BioXcell | BE0017-1-50MG | InVivoMAb anti-mouse CD40L (CD154) | 50 mg |
| BioXcell | BE0017-1-100MG | InVivoMAb anti-mouse CD40L (CD154) | 100 mg |
| BioXcell | BE0019-1MG | InVivoMAb anti-mouse CD45RB | 1 mg |
| BioXcell | BE0019-5MG | InVivoMAb anti-mouse CD45RB | 5 mg |
| BioXcell | BE0019-25MG | InVivoMAb anti-mouse CD45RB | 25 mg |
| BioXcell | BE0019-50MG | InVivoMAb anti-mouse CD45RB | 50 mg |
| BioXcell | BE0019-100MG | InVivoMAb anti-mouse CD45RB | 100 mg |
| BioXcell | BE0019-1-1MG | InVivoMAb anti-human CD47 | 1 mg |
| BioXcell | BE0019-1-5MG | InVivoMAb anti-human CD47 | 5 mg |
| BioXcell | BE0019-1-25MG | InVivoMAb anti-human CD47 | 25 mg |
| BioXcell | BE0019-1-50MG | InVivoMAb anti-human CD47 | 50 mg |
| BioXcell | BE0019-1-100MG | InVivoMAb anti-human CD47 | 100 mg |
| BioXcell | BE0020-1-1MG | InVivoMAb anti-mouse CD54 (ICAM-1) | 1 mg |
| BioXcell | BE0020-1-5MG | InVivoMAb anti-mouse CD54 (ICAM-1) | 5 mg |
| BioXcell | BE0020-1-25MG | InVivoMAb anti-mouse CD54 (ICAM-1) | 25 mg |
| BioXcell | BE0020-1-50MG | InVivoMAb anti-mouse CD54 (ICAM-1) | 50 mg |
| BioXcell | BE0020-1-100MG | InVivoMAb anti-mouse CD54 (ICAM-1) | 100 mg |
| BioXcell | BE0020-2-1MG | InVivoMAb anti-human CD54 (ICAM-1) | 1 mg |
| BioXcell | BE0020-2-5MG | InVivoMAb anti-human CD54 (ICAM-1) | 5 mg |
| BioXcell | BE0020-2-25MG | InVivoMAb anti-human CD54 (ICAM-1) | 25 mg |
| BioXcell | BE0020-2-50MG | InVivoMAb anti-human CD54 (ICAM-1) | 50 mg |
| BioXcell | BE0020-2-100MG | InVivoMAb anti-human CD54 (ICAM-1) | 100 mg |
| BioXcell | BE0021-1MG | InVivoMAb anti-mouse L-Selectin (CD62L) | 1 mg |
| BioXcell | BE0021-5MG | InVivoMAb anti-mouse L-Selectin (CD62L) | 5 mg |
| BioXcell | BE0021-25MG | InVivoMAb anti-mouse L-Selectin (CD62L) | 25 mg |
| BioXcell | BE0021-50MG | InVivoMAb anti-mouse L-Selectin (CD62L) | 50 mg |
| BioXcell | BE0021-100MG | InVivoMAb anti-mouse L-Selectin (CD62L) | 100 mg |
| BioXcell |
风险提示:丁香通仅作为第三方平台,为商家信息发布提供平台空间。用户咨询产品时请注意保护个人信息及财产安全,合理判断,谨慎选购商品,商家和用户对交易行为负责。对于医疗器械类产品,请先查证核实企业经营资质和医疗器械产品注册证情况。
 询价记录
询价记录
 技术资料
技术资料
需要更多技术资料 索取更多技术资料
BioXCell——CD Marker抗体.pdf 附 (下载 0 次)


