大家都在搜


确定一种蛋白质的结构和功能是现代生物学许多研究的基础。在过去的几十年里,人们开发了许多用于结构预测的计算工具,其中 Phyre2 因其强大的分析能力与简单的操作被生物科研人员广泛使用,据统计每天都有约 1000 个任务提交到 Phyre2 服务器上。
Phyre2 分析能力强大,结果界面丰富直观,可以预测和分析蛋白质的结构、功能和突变。相比之前的版本 Phyre v 1.0,Phyre2 使用更先进的远程同源检测方法来构建蛋白质三维模型,预测配体结合位点,并分析氨基酸突变(如 SNPs)对目标蛋白序列的影响。接下来让我们一起来感受下。
登录 Phyre2 主页 (http://www.sbg.bio.ic.ac.uk/phyre2),复制并粘贴目标氨基酸序列到框中。填写提醒邮箱。选择 Normal(普通)或 Intensive(强化)模式。提交任务后页面会显示任务进度和预测所需时间。
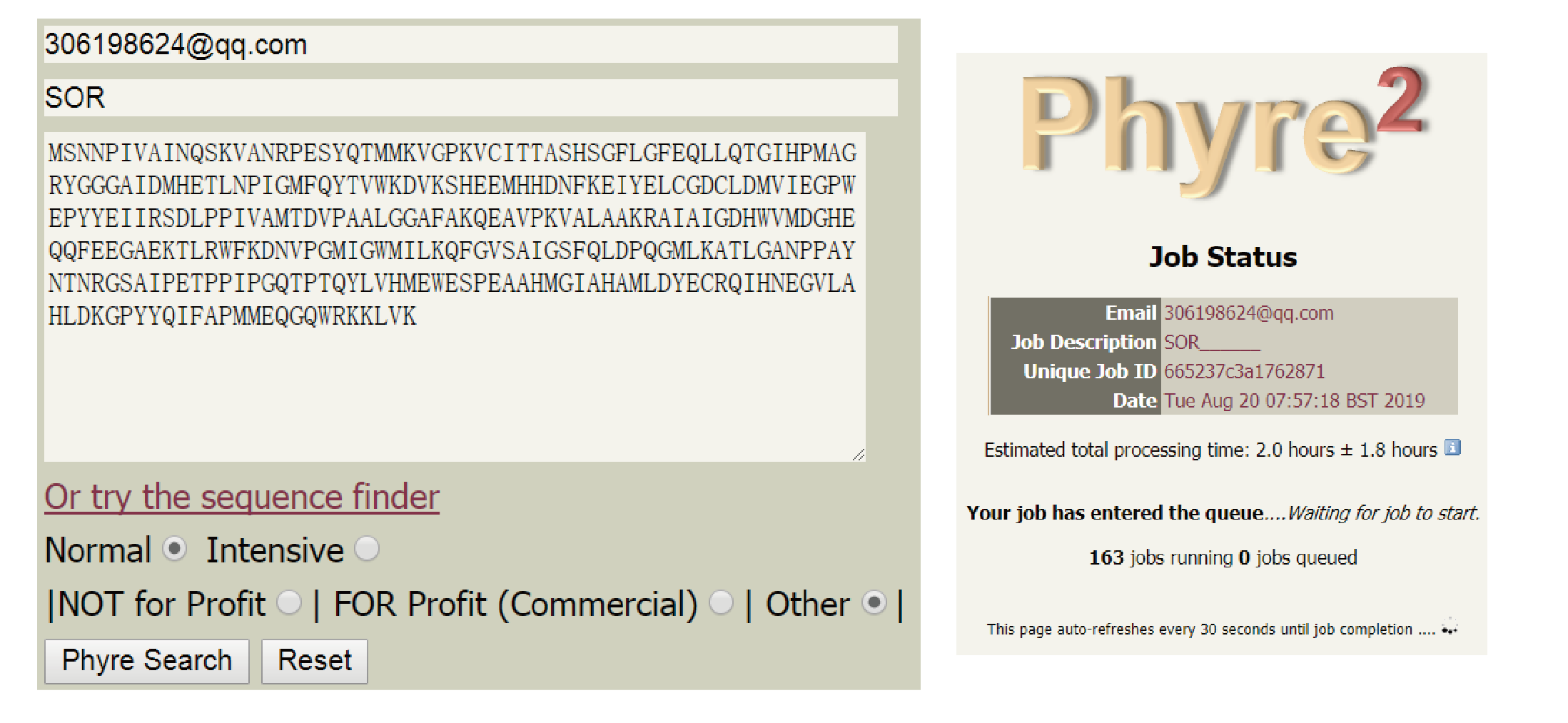
提交 Phyre2 任务
结果页面
页面分为几个主要部分:总结、序列分析、二级结构预测、结构域分析和模板的详细信息。在某些情况下结合位点预测和跨膜螺旋的预测。
1. 序列分析:点击「查看 PSI-Blast 多序列比对」按钮会打开一个新窗口,包含使用 PSI-Blast 对最新的非冗余蛋白序列库扫描查询序列的结果。为了确保比对能产生准确的二级结构预测,同源蛋白评估的 E 值应<0.001。
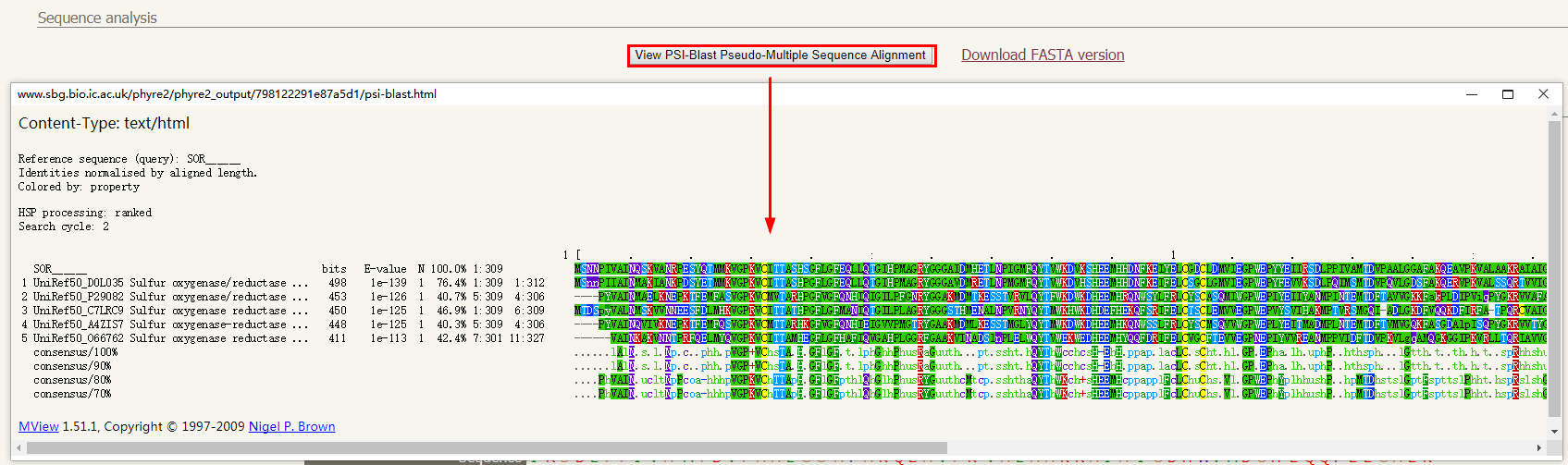
序列分析
2. 二级结构(α-helix, β-strand 或 coil)的预测:模块的第一行显示了序列的位置。残基基于氨基酸性质进行着色:极性的氨基酸 A,S,T,G,P 为黄色,疏水性的氨基酸 M,I,L,V 为绿色,荷电性的氨基酸 K,R,E,N,D,H,Q 为红色,芳香性的氨基酸 W,Y,F,C 为紫色。置信度线评估区域预测的可信度,红色表示高置信度,蓝色为低置信度。只有在序列数据库中检测到大量不同的序列同源序列时才能达到 78 - 80% 的准确度。一般不考虑置信度 < 90% 的模型。
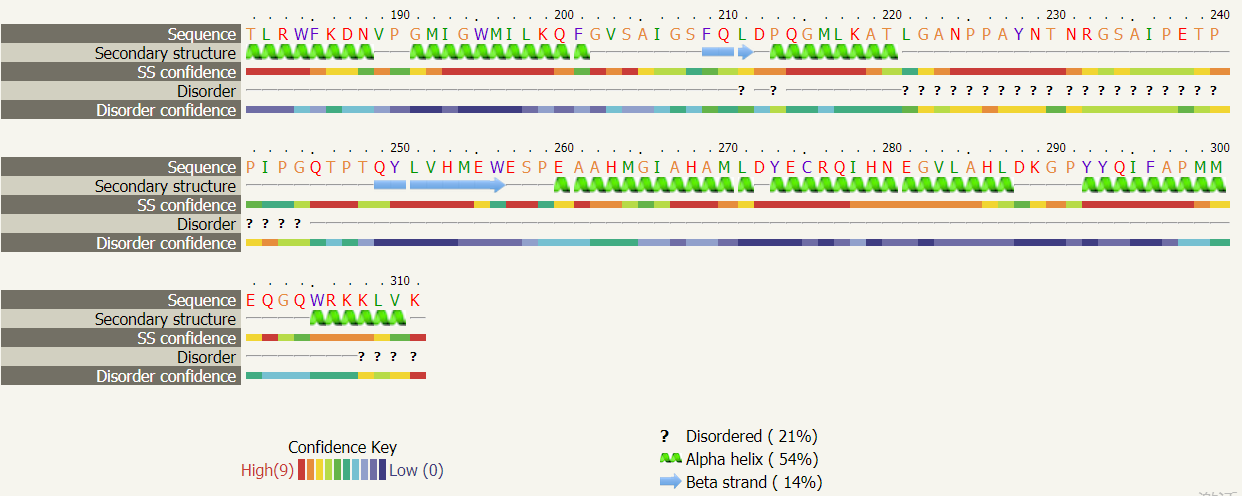
二级结构类型的预测
3. 结构域分析:通过 Phyre2 与已知结构序列的匹配,上传序列和已知序列结构之间对齐,框的宽度表示比对上的长度用颜色表示对同源性的置信度(红色表示高置信度,蓝色为低置信度)。将鼠标悬停在它们上面,会弹出模型的图片和详细信息。单击此链接可转到详细结果表中对应的条目。能够看到上传序列是否有一些区域是无法建模,或者是否有多个模板覆盖上传序列的不同区域,指示上传序列可能的蛋白质结构域。
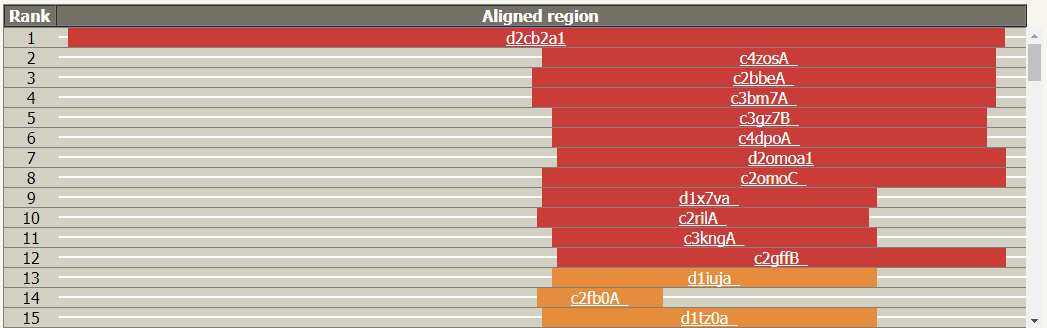
结构域分析
4. 详细模板信息表:显示使用的 PDB 模板及其代码,从 PDB 文件中提取关于蛋白质模板的信息、对模型的置信度和上传序列的覆盖率、3D 模型、置信度、百分比序列标识和文本描述。在这一栏中还有一个名为的按钮。如果对匹配的模型特别感兴趣,可以通过单击「Phyre Investigator」在弹出的页面中点击 submit 按钮提交。
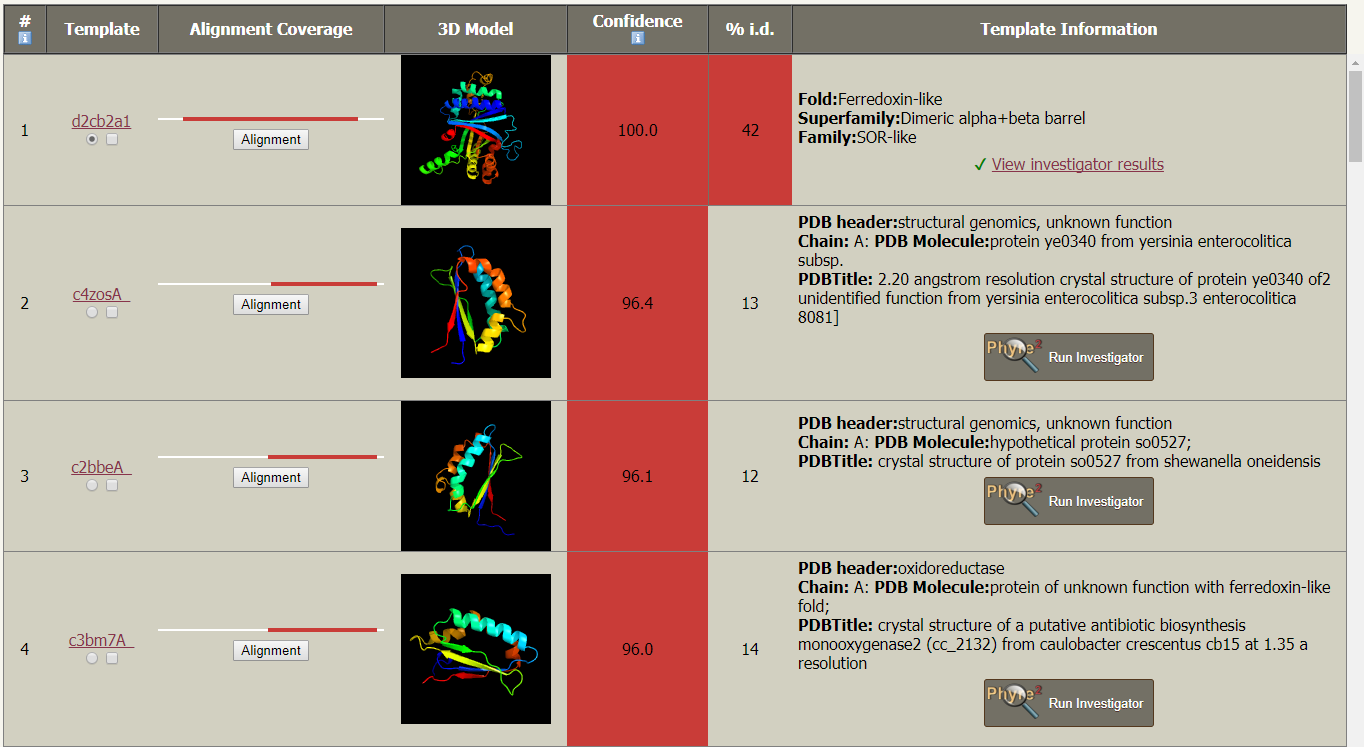
模板信息
5. 使用 Phyre Investigator 深入分析:Phyre Investigator 界面由几个部分组成,点击「Analysis」部分中的「Quality」选项卡,可以看到包括 ProQ2(模型质量预测系统)、clashes、rotamers 和 Ramachandran 分析等在内的选项按钮。点击这些按钮可以比较模型区域的质量。如果建模质量低的残基与您要研究的残基相距很远,或者处于在功能上不太重要的环(loop),则不需要考虑它们。但是,如果质量低的残基位于功能位点附近,则应谨慎考虑,并研究其他可能避免这些问题的替代模型或方法。单击 Analyses(分析)部分的 Function(函数)选项卡,可以显示保守区域、接触界面、口袋和突变敏感区等选项。保守区域可以为查找功能残基提供线索。如果高度保守的残基恰好存在于口袋中,这就能确切说明其功能是十分重要的。
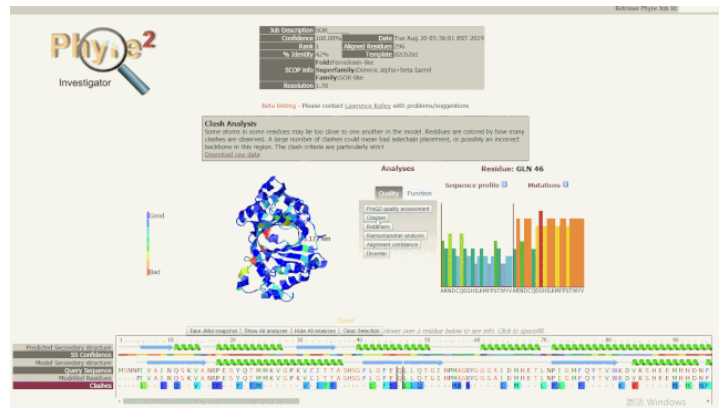
使用 Phyre Investigator 查看模型质量
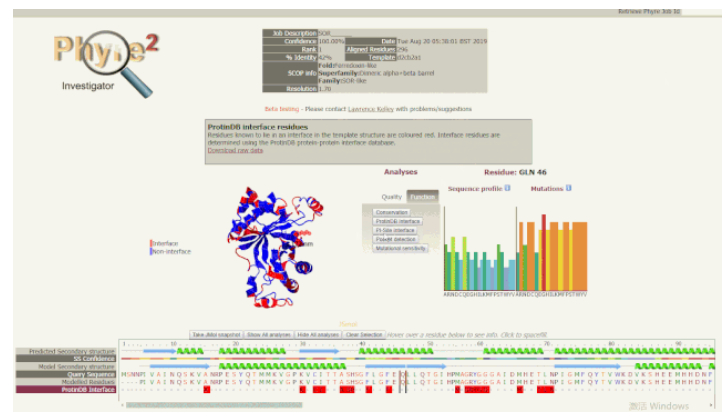
使用 Phyre Investigator 查看蛋白功能
随着光标在序列视图中移动,模型上的红色指示点以及右侧的序列谱(Sequence Profile)和突变倾向图(Mutations)也会随之移动或改变。如果某些残基被预测为质量较低,但又可能具有重要的功能,可以在序列视图中悬停它,查看它们的突变情况和突变图示。进而可以探究一下突变是否对某些类型的氨基酸有强烈的偏好,某些突变敏感度高的残基可能具有显著表型效应,而这些信息可以指导进一步的突变实验,帮助解释单核苷酸多态性(SNPs)的作用。Phyre2 具有预测点突变表型效应的功能,但不能准确地确定点突变在超出侧链估计位置之外结构上的影响。
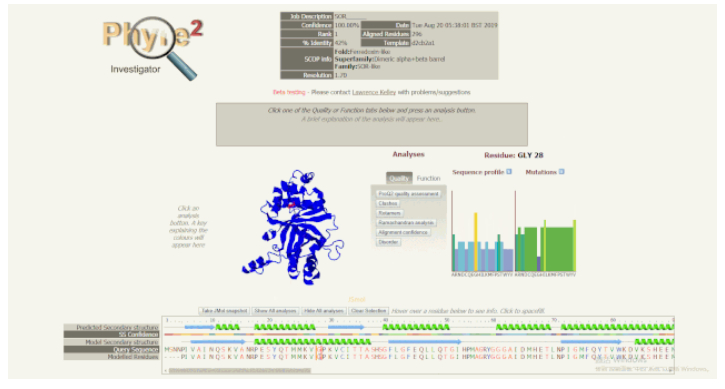
序列视图的使用
6. 模型叠加分析(Superposition)
在选择一个主模型之后,还可以勾选多个从模型来进行叠加。点击主表的底部 Generate superposition of selected models(生成所选模型的叠加)的按钮。叠加使用 MaxSub 算法 m33 执行。该算法能够找出在 4.5A 范围内叠加的两个结构之间原子的最大子集。一般情况下,会选择得分最高的模型作为主模型,或者根据之前的一些生物学知识背景来判断值得研究的模型。

生成所选模型的叠加
点击表格下方的 Generate superposition of selected models(生成所选模型的叠加)的按钮,选中的模板将叠加在主模型上。进入的页面中有一个 JSMol 窗口,用于交互查看叠加的模型,右侧有相应的一些描述和帮助信息。其中 RMSD(均方根偏差)显示了叠加构象与目标构象所有原子偏差的加和,其值越小,构象差异越小。手动旋转叠加的模型,在三维空间中可以寻找模型的哪些区域是一致的。通常可以观察到一个保守内核(core)具有可变的表面环(surface loop),它可以表明哪里可能存在建模错误或具有结构灵活性。
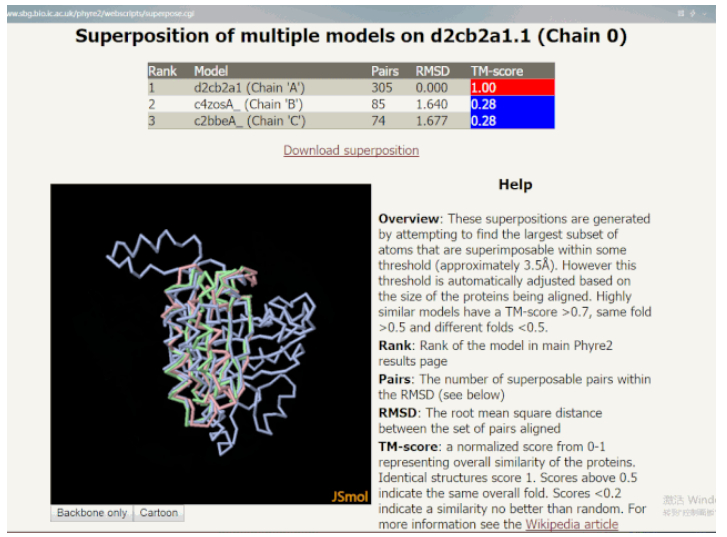
交互查看叠加的模型
7. 结合位点预测:如果给得分最高的模型预测出的概率 >90%,则模型和序列会自动提交给 3DLigandSite 服务器进行配体结合位点预测。蛋白质通常作用于配体 (如酶底物) 上或受配体调控。3DLigandSite 将类似结构的配体叠加到目标蛋白的结构模型上,并将残基保存映射到蛋白质表面,结合两种方法产生的数据进行预测。3DLigandSite 的结果分为四个主要部分。第一个提供了使用的 phyre 模型信息。第二部分显示了已识别的配体簇表。后两个部分显示 3DLigandSite 预测。表中列出了所有预测的结合位点残基,详细说明了它们所接触的配体数量、残基与残基保存分数 (JSD) 之间的平均距离。
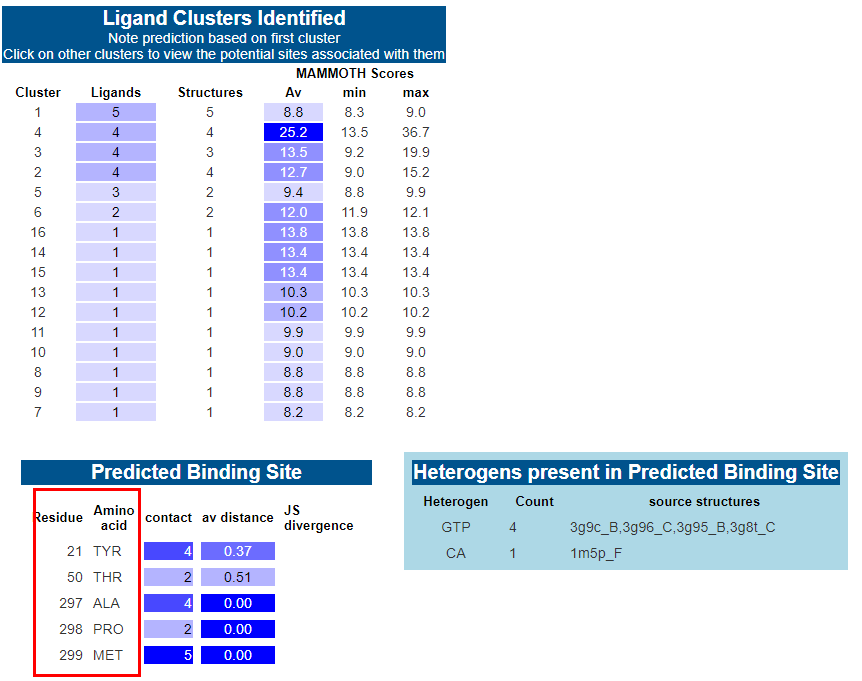
结合位点预测
Jmol 对建模的蛋白质、配体集群和预测的结合位点进行了可视化。用户还可以对预测残基进行标记,以便研究预测的结合位点。
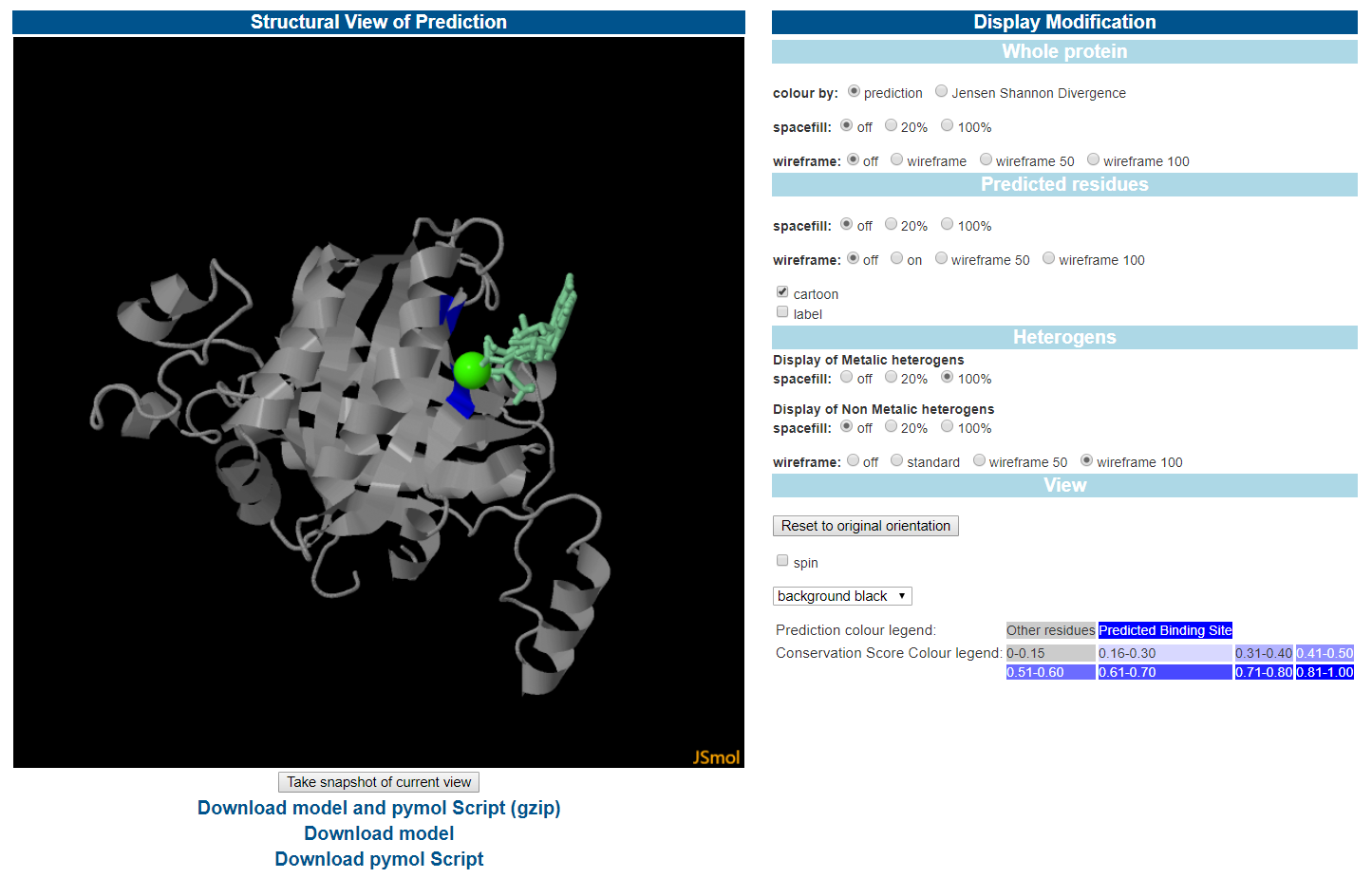
结合位点可视化
8. 跨膜螺旋的预测:Phyre2 通过支持向量机(一种机器学习工具)处理上传的序列和 PSI-Blast 检测到的同系序列,会确定上传序列是否包含跨膜螺旋,并预测它们在膜中的拓扑结构(平均准确率为 89%)。如果预测跨膜螺旋,将显示每个跨膜螺旋的开始和结束以及膜的胞外和胞质侧示意图,并用数字表示残留指数。
此外,在结果页面顶部有一个包含基本信息及有效期的框。如果任务即将到期,文本变成红色,这时点击「续领 30 天」链接,可以重置结果的有效期。也可以将结果全部下载保存(点击 Download zip of all results)。
任务信息
文中有动图,具体可参考:

更有优质直播、研选好物、福利活动等你来!


我的询价
询价列表

暂时没有已询价产品
手机验证