- 移动端


上海申知心生物科技有限公司
2 年
手机商铺
- NaN
- 0
- 0
- 0
- 0
技术资料/正文
NET-DNA-CCDC25轴驱动乳腺癌化疗/放疗耐药的机制与干预策略
170 人阅读发布时间:2026-01-23 13:55

² 研究背景
乳腺癌是女性癌症死亡的首要病因,手术联合化疗/放疗是标准治疗方案,但治疗后耐药频发且机制未明。细胞毒疗法在杀伤肿瘤细胞的同时,会释放大量损伤相关分子模式(DAMPs),这些危险信号虽可激活抗肿瘤免疫,却也常诱导免疫抑制微环境,最终削弱疗效。近年来,中性粒细胞胞外诱捕网(NET)及其DNA成分(NET-DNA)被证实参与肿瘤进展,但其在化疗/放疗抵抗中的功能角色、临床意义及上下游调控机制仍不清楚。
现有靶向NET-DNA的策略(如DNase I降解)虽可抑制转移,但存在非特异性切割DNA、损害宿主免疫防御等局限。因此,亟需明确治疗诱导NET-DNA促进耐药的精确机制,并识别可干预的关键节点。本研究假设NET-DNA通过特异性受体激活癌细胞上皮-间质转化(EMT)程序从而降低治疗敏感性,旨在阐明NET形成的上游驱动信号及癌细胞感知NET-DNA的下游效应通路,为开发精准、低毒的联合干预策略提供理论依据。
² 研究结果
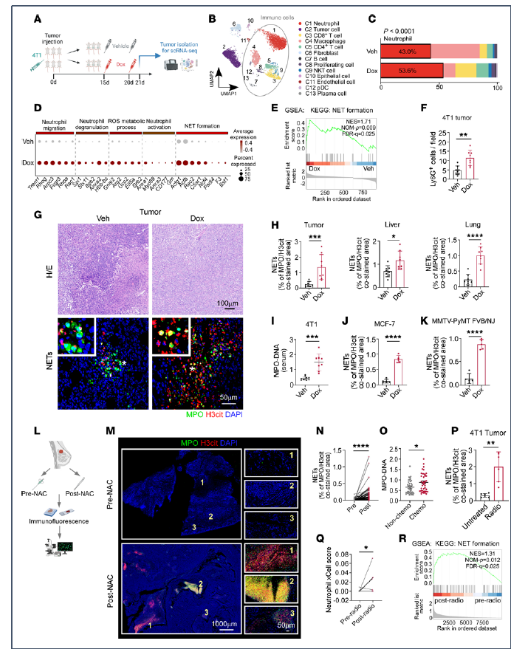
1. 化疗和放疗促进乳腺癌中NET的生成
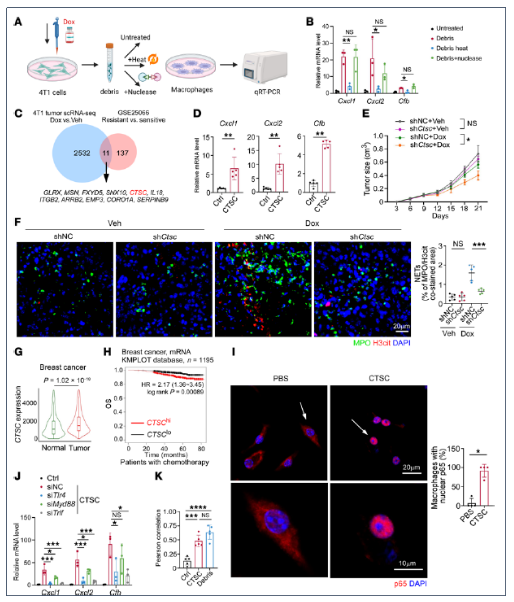
研究者首先对4T1移植瘤进行scRNA-Seq,发现多柔比星(DOX)处理后瘤内中性粒细胞比例显著升高,其转录特征显示NET形成通路(NETosis)富集明显。免疫荧光进一步验证化疗组瘤内Ly6G+中性粒细胞浸润增加,且H3cit与MPO共定位的NET结构增多,血清MPO-DNA复合物水平同步上升。该现象具有普适性:在激素受体阳性MCF-7模型、HER2富集型MMTV-PyMT自发瘤模型以及接受新辅助化疗的81例患者配对活检标本中,治疗后NET表达均显著上调。放疗同样提升瘤内NET水平,且乳腺癌患者放疗后转录组数据及Lewis肺癌公共数据均显示中性粒细胞评分与NET通路增强。综上,化疗/放疗通过激活NETosis程序,在原发瘤、转移灶及外周血中系统性增加NET负荷。
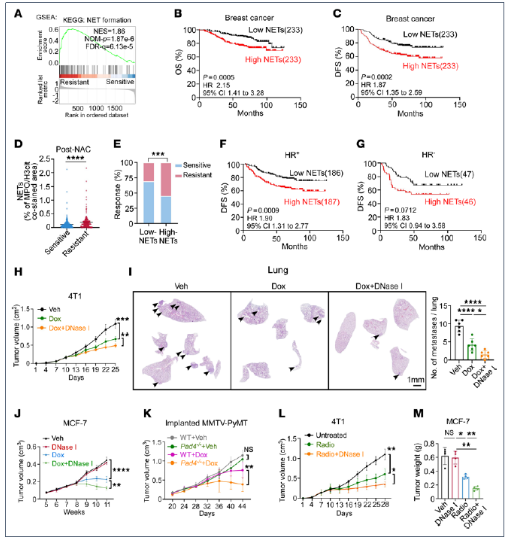
2.NET生成增强削弱化疗和放疗疗效
通过公共数据集GSEA分析,耐药肿瘤中NET形成通路显著上调;对233例接受新辅助化疗(NAC)的患者队列进行Kaplan-Meier分析,高NET组总生存期(OS)和无病生存期(DFS)均显著缩短,多因素Cox回归确认其为独立预后因子。功能上,DNase I降解NET-DNA或利用Pad4⁻/⁻小鼠(NET生成缺陷)联合多柔比星/放疗,使4T1和MCF-7模型肿瘤体积较单药组再缩小40–60%,肺肝转移灶减少>50%,凋亡指数提升2倍以上。临床转化层面,高NET患者对NAC客观反应率降低,且在HR+亚型中预后价值更突出。这些数据证实NET不仅是耐药标志物,更是可直接干预的功能性靶点。
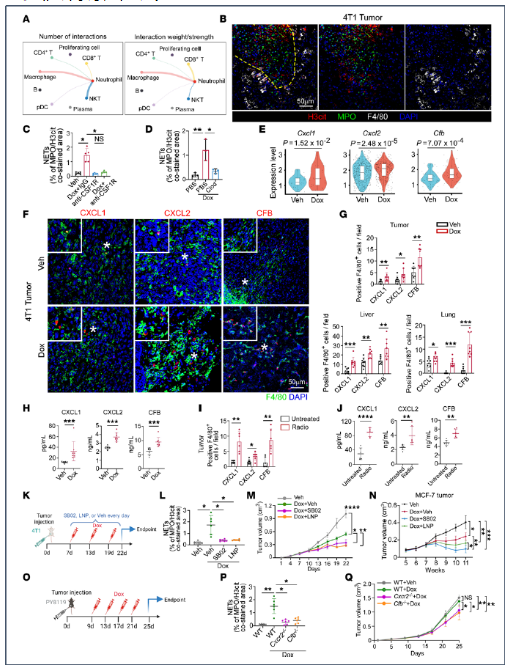
3. 巨噬细胞来源的CXCL1/2和CFB在化疗/放疗下促进中性粒细胞浸润与NETosis
scRNA-Seq细胞通讯分析显示巨噬细胞与中性粒细胞互作权重最强,空间定位显示NET聚集于巨噬细胞富集区。清除巨噬细胞(抗CSF1R抗体或氯膦酸盐)后,多柔比星诱导的Ly6G+细胞浸润和NET生成减少60%以上。机制上,化疗后巨噬细胞CXCL1/2与补体因子B(CFB)表达上调3–5倍,CXCR2抑制剂SB225002或CFB抑制剂LNP023联用使瘤内NET下降55%和48%,肿瘤生长分别再抑制42%与38%。Cxcr2⁻/⁻和Cfb⁻/⁻基因敲除小鼠进一步验证该通路必要性,且抑制该轴可减少肺/肝转移灶形成。因此,巨噬细胞通过分泌CXCL1/2和CFB,构成治疗诱导NET扩增的关键驱动环路。
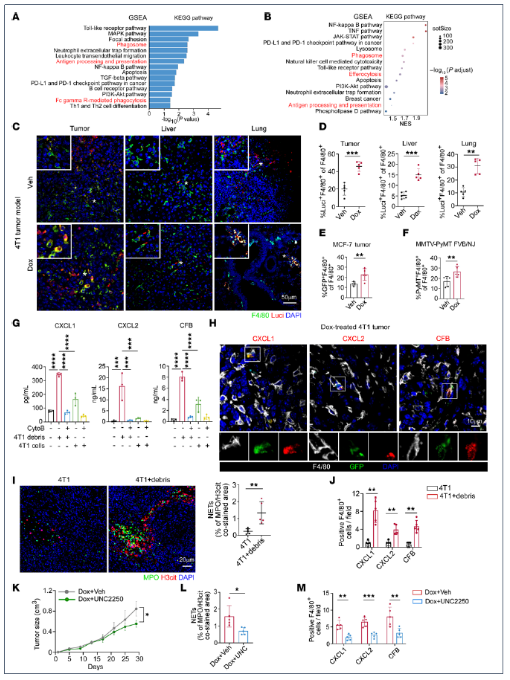
4. 巨噬细胞通过吞噬治疗诱导的肿瘤碎片促进CXCL1/2和CFB表达及NETosis
scRNA-Seq显示化疗后巨噬细胞“吞噬体”和“FcγR介导的吞噬”通路富集,体内成像证实Luci⁺/GFP⁺/PyMT⁺肿瘤碎片被F4/80⁺巨噬细胞吞噬比例增加2–3倍。体外实验表明,肿瘤碎片刺激使骨髓来源巨噬细胞(BMDM)分泌CXCL1/2和CFB提升4–6倍,而细胞松弛素B(CytoB)抑制肌动蛋白聚合或MerTK抑制剂UNC2250阻断吞噬后,该效应被逆转70%以上。体内联合UNC2250与化疗,使瘤内吞噬碎片巨噬细胞减少50%,NET密度同步下降,肿瘤生长延缓。将肿瘤碎片与活细胞共注射可增强NET形成,证实碎片是激活巨噬细胞炎症程序的危险信号,从而桥接治疗损伤与NET扩增。
5. 肿瘤碎片中的CTSC蛋白在化疗后促进NETosis
通过交叉筛选耐药肿瘤与scRNA-Seq数据,发现CTSC在两者中均上调;敲低CTSC使碎片诱导的炎症因子表达降低60%以上。机制上,重组CTSC蛋白直接刺激巨噬细胞,通过TLR4/TRIF(而非MyD88)途径促进p65核转位,激活NF-κB。TLR4与RAB5在早期内体共定位,证实内体TLR4识别CTSC。临床数据支持其相关性:乳腺癌瘤内CTSC表达高于正常组织,且高CTSC患者接受化疗后生存期缩短。体内敲低肿瘤细胞CTSC联合化疗,使瘤体积缩小55%,NET密度下降50%,从而确立CTSC为治疗损伤感知与NET扩增的关键介质。
6. NET通过CCDC25促进乳腺癌治疗抵抗
临床队列分析显示,瘤内CCDC25高表达与HR+患者NAC后DFS/OS缩短及病理wan全缓解率降低相关。体外NET刺激使CCDC25⁺的MCF-7和T47D细胞对多柔比星IC50升高,E-cadherin下降,Vimentin/N-cadherin增加,而敲除或中和CCDC25可逆转EMT并恢复药物敏感性。体内实验证实,CCDC25敲除的MCF-7移植瘤或4T1模型在化疗/放疗下肿瘤体积分别缩小70%和50%,凋亡指数提升2.5倍,但瘤内NET总量无变化,表明CCDC25作用位于NET下游。MMTV-PyMT模型中,抗CCDC25中和抗体联合化疗显著抑制肿瘤生长及肺肝转移,为靶向该受体提供直接证据。

7. NET-CCDC25相互作用通过STAT3激活和EMT促进肿瘤细胞耐药
RNA-Seq显示NET处理后MCF-7细胞EMT通路富集度居首,且STAT3靶基因集同步上调。Pull-down/质谱鉴定PKM2为CCDC25结合蛋白;NET刺激后PKM2核转位增加,与STAT3 Tyr705磷酸化及TWIST1、SNAIL启动子结合呈正相关。CCDC25敲除使STAT3核结合能力下降。功能上,敲低STAT3或使用抑制剂Stattic可阻断NET诱导的EMT标志物转换,并恢复MCF-7对多柔比星敏感性。PKM2抑制剂Shikonin阻止其核定位,也抑制STAT3磷酸化及EMT。因此,NET-DNA结合CCDC25后招募PKM2入核,进而磷酸化STAT3,驱动EMT转录程序,形成完整的“NET-CCDC25-PKM2-STAT3-EMT”耐药轴。

² 研究结论
化疗和放疗通过“肿瘤碎片→巨噬细胞吞噬CTSC→TLR4/TRIF/NF-κB→ CXCL1/2/CFB”信号级联,显著增加瘤内及转移灶的NET形成。释放的NET-DNA与癌细胞膜受体CCDC25结合,经PKM2介导STAT3磷酸化,激活EMT程序,导致乳腺癌尤其是HR+亚型对治疗耐受。靶向阻断CCDC25、CXCR2或CFB可在保留宿主免疫防御的同时,显著增强化疗/放疗疗效,为临床克服耐药提供了新的组合策略和可干预的生物标志物。
