- 移动端


上海生物芯片有限公司
3 年
手机商铺
- NaN
- 0.7999999999999998
- 0.7999999999999998
- 1.7999999999999998
- 0.7999999999999998
推荐产品




公司新闻/正文
肿瘤突变特征推断工具deconstructSigs
558 人阅读发布时间:2024-07-09 14:05
我们知道组成DNA序列的碱基总共有四种(A、T、C、G),而点突变(SNV)就是这四种碱基之间的替换,可以计算出替换类型为4*3=12种,考虑到碱基互补配对,我们将其归纳为六种突变类型:C>A/G>T,C>G/G>C,C>T/G>A,T>A/A>T,T>C/A>G,T>G/A>C。但仅仅考虑单个位点的突变难以形容生物体中复杂的功能,而功能基本单位氨基酸对应DNA序列上的密码子,密码子由三联碱基组成,根据点突变(SNV)碱基种类以及其上、下游 1bp 碱基种类,可以将点突变归纳分为 96 种(4x6x4)突变类型,我们把这96种碱基替换类型作为一种特征进行研究,以分析研究癌症的点突变类型的偏好和各种癌样本在点突变水平上的相似程度。
COSMIC统计了不同的肿瘤突变特征,并分析了各种突变特征的意义。

今天我们要介绍一种已知肿瘤突变特征推断工具deconstructSigs,这是一个R包,通过比非负矩阵分解(NMF),将样本突变特征映射到特征库中已知的特征矩阵上,从而获取该样本存在哪些特征,查询COSMIC网站获取特征含义。
简单说一下NMF原理:该算法同时输入 n 个样本突变频率的96*n阶矩阵V,以及k种已知特征突变频率的96*k阶矩阵 S,基于 T 和 S 的最小误差平方和 SSE 选取最能代表样本信息的突变特征,重构矩阵R,并依次进行迭代,直至前后两次迭代的 SSE<0.001 为止,最终得到每种突变特征的权重矩阵W。
PS:理解不了的可略过,下面开始讲重点!
首先是软件的下载安装,这里推荐用conda安装,简单方便。支持新版本R,小编就是在R 4.3.3下安装的。
# 安装R包
conda install r-deconstructSigs
# 安装基因组文件
BiocInstaller::biocLite('BSgenome')
BiocInstaller::biocLite('BSgenome.Hsapiens.UCSC.hg19')
PS:这个基因组文件后面用于构建96种突变特征矩阵,deconstructSigs提供了转换函数,当然也可以自己根据参考基因组构建。
# 安装完成后导入
library("deconstructSigs")
先不急,我们看看R包内置的Signature数据库,软件内置了两套signature数据库,逐个打开看看:
# 查看signatures.nature2013数据库
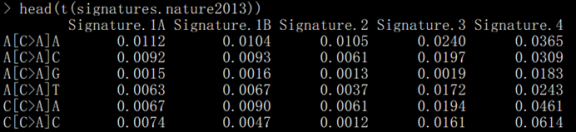
head(t(signatures.nature2013))
# 查看特征数
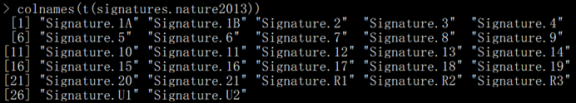
colnames(t(signatures.nature2013))
# 查看signatures.cosmic数据库
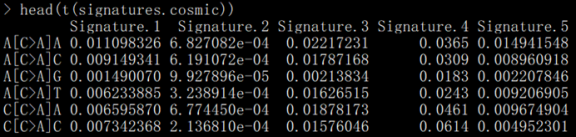
head(t(signatures.cosmic))
# 查看特征数
colnames(t(signatures.cosmic))
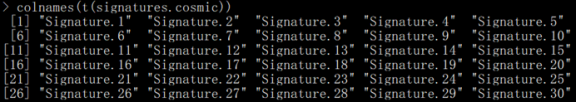
可以看到每列对应一个signature,行数对应96种突变情况,中间的数值是每一个signature的96种突变情况的占比(每列和为1);signatures.nature2013数据有27种signature,而signatures.cosmic数据有30种sginature,分析时可以选择某一个,软件默认用signatures.nature2013数据。接下来,开始制作输入文件。通过肿瘤突变检测可以得到每个肿瘤样本的体细胞突变文件,如果是VCF文件,最好先进行注释,因为我们要根据注释信息进行突变位点筛选,只考虑对氨基酸有影响的点突变(SNV),这里用的是ANNOVAR注释结果,筛选标准如下:
(1)筛选Filter列,保留”PASS”
(2)筛选突变区域,保留”exonic”、”splicing”、“exonic;splicing”
(3)筛选非同义突变,去除“.”、”synonymous SNV”、”unknown”
(4)筛选DP>=20、Nalt>=3、AF>0.01或0.05
(5)只保留snv
提取筛选后的突变结果信息,需要五列:Sample、chr、pos、ref、alt,如下所示:
## Sample chr pos ref alt
## T1_vs_N1 chr10 39948943 C T
## T1_vs_N1 chr10 41524064 T C
## T1_vs_N1 chr10 61107628 C T
把该文件作为输入文件,通过mut.to.sigs.input转换为96种突变特征矩阵。
# Using deconstructSigs
sample.mut.ref <- read.table("T1.somatic.tsv", header = FALSE, sep = "\t")
head(sample.mut.ref)
# mutfile, tsv format. ucsc.hg19
sigs.input <- mut.to.sigs.input(mut.ref = sample.mut.ref,
sample.id = "T1_vs_N1",
chr = "chr",
pos = "pos",
ref = "ref",
alt = "alt",
bsg = BSgenome.Hsapiens.UCSC.hg19)
PS:这里的sigs.input就是样本96种突变特征矩阵,可以自己制作,需要注意表头碱基替换类型保持一致。
我们可以将多个样本的sigs.input矩阵合并为一个,当然也可以不合并,文件格式如下:
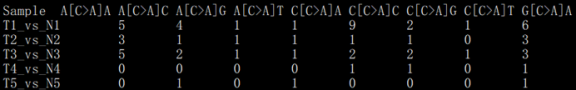
现在我们可以进行样本signature推断了,使用whichSignatures函数:
sample.tumors = whichSignatures(tumor.ref = sigs.input,
signatures.ref = signatures.cosmic,
sample.id = sample,
contexts.needed = TRUE,
tri.counts.method = 'default')
参数说明:
tumor.ref:每个sample的96种三碱基突变序列signatures.ref:已知的signatures参考文件,可选signatures.nature2013和signatures.cosmicsample.id:对应tumor.ref文件中的样本名,每次只能运行一个样本contexts.needed :是否需要突变上下文tri.counts.method:三核酸序列标准化方式,默认“default” 不进行标准化 ;或者选择exome,genome,exome2genome,genome2exome 来限定区域。
whichSignatures会输出5个元素的list文件:
# 查看结果结构
class(sample.tumors)
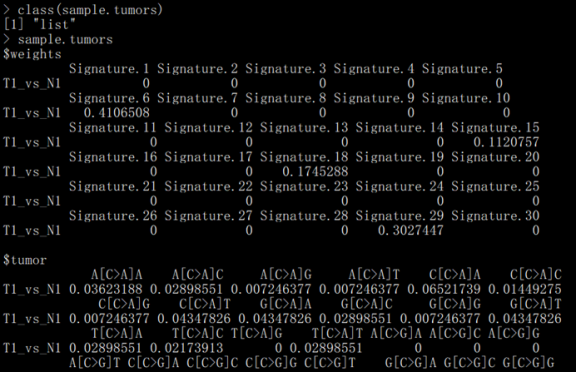
weights:包含分配给输入签名矩阵的每个k个签名的权重的数据帧
tumor:作为输入的肿瘤样本的三核苷酸背景矩阵
product:当肿瘤矩阵乘以指定的权重时得到的矩阵
diff:表示肿瘤矩阵与乘积矩阵之差的矩阵
unknown:未分配给任何输入签名的数字权重
最后,为了更直观了解样本signature情况,使用plotSignatures函数可视化。
# 绘制突变特征推断结果
plotSignatures(sample.tumors)
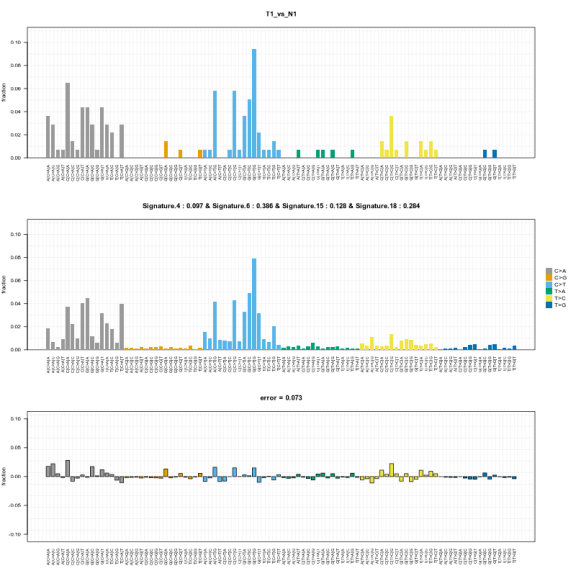
样本突变特征图
上图为该样本突变模式实际分布图,中图为由已知 cosmic signature构建出来的突变模式图,下图为两者的 sum-squared error,值越低越少。
# 绘制突变特征权重饼图
makePie(sample.tumors)
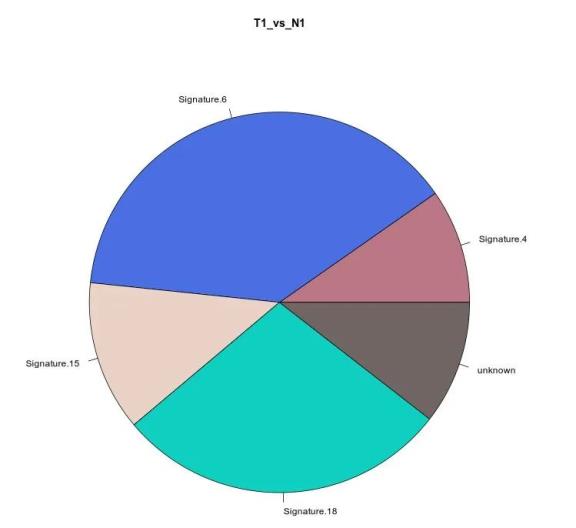
样本突变特征权重饼图
这里是根据已知signature进行推断,那未知的signature是如何推断的呢?下期再给大家分享。