- 移动端


上海生物芯片有限公司
3 年
手机商铺
- NaN
- 0.7000000000000002
- 0.7000000000000002
- 1.7000000000000002
- 0.7000000000000002
推荐产品




公司新闻/正文
单细胞分析雕刻师--常见整合方法比较(三)
2083 人阅读发布时间:2023-06-07 10:27
Preface
随着单细胞测序技术的迅猛发展和市场的不断下沉,越来越多的研究人员都青睐于使用该技术来阐明一些生物学或医学问题,使通过传统bulk-RNA测序无法解决的事情得以实现成为可能,如细胞图谱的绘制、稀有细胞的鉴定与识别、细胞发育/分化轨迹的构建、肿瘤的精细化研究等。与此同时,也产生了海量的单细胞数据,而这些数据通常来源于不同的实验室,具有不同的构建时间、不同的操作人员以及不同的试剂批次等等。上述差异往往会对数据的合并造成严重的影响,导致批次效应的出现,进而干扰对真实的生物学效应的鉴别,因此,如何将不同来源的数据完美地系在一起一直是一个复杂的、具有挑战性的问题。在过去的十几年间,有数十种数据整合方法相继被开发出来,它们基于不同的原理或应用场景实现对数据的合并,在保留生物学差异的同时尽可能地去除批次效应。这里,我们选择了一些比较常见的工具或方法,包含ComBat、BBKNN、Seurat CCA、Seurat RPCA、Harmony、LIGER、fastMNN、Conos、Scanorama总共9种,通过应用于同一套数据对其进行比较。
书接上回,《单细胞分析雕刻师--常见整合方法比较(一)》与《单细胞分析雕刻师--常见整合方法比较(二)》分别为大家带来了6类常见的整合方法,本期推送继续为大家带来最后3类常见的整合方法。
Results
07 ComBat
ComBat[7]最初常用于bulkRNA、芯片表达数据的批次矫正。该方法主要依赖经验贝叶斯估计批次效应,从而修正并返回一个新的表达矩阵。因为Scanpy内部直接了整合该方法,因此我们在Python环境下执行本次分析。通过函数 scanpy.pp.combat 对数据进行整合。

Figure15 | ComBat 整合分析结果。左边为UMAP降维图形展示,分别以数据集和细胞类型分组;右图是结果评分。
08 Conos
Conos (clustering on network of samples) 方法侧重于异质样品集合中同源细胞类型的统一映射,通过比较不同数据集的所有细胞配对情况来构建联合图(Figure16)。为了实现这一目的,Conos会执行配对比较,识别合理的样本间细胞关系[8]。但由于样本间的差异并不总是能被很好地模拟,单个的比对比较容易出错,然而,在许多成对比较中,相似的细胞亚群将倾向于相互映射,形成一个小的集合。所以Conos会综合比对结果,从而建立样本间及细胞间的加权连接。Conos的处理可大致分为三个阶段:1)过滤和标准化:对每个数据集进行数据处理,包括低质量细胞的过滤、文库标准化等,依赖于第三方包如pagoda2或Seurat;2)确认样本间匹配:在降维空间中执行数据集间的配对比较,初步建立比对网;3)联合图构建:结合样本间及样本内连接构成联合图。这里,我们主要通过函数 conos::buildGraph(space = "PCA", matching.method = "mNN") 来完成图形构建,以及 conos::embedGraph(method = "UMAP") 建立联合图的二维嵌入。
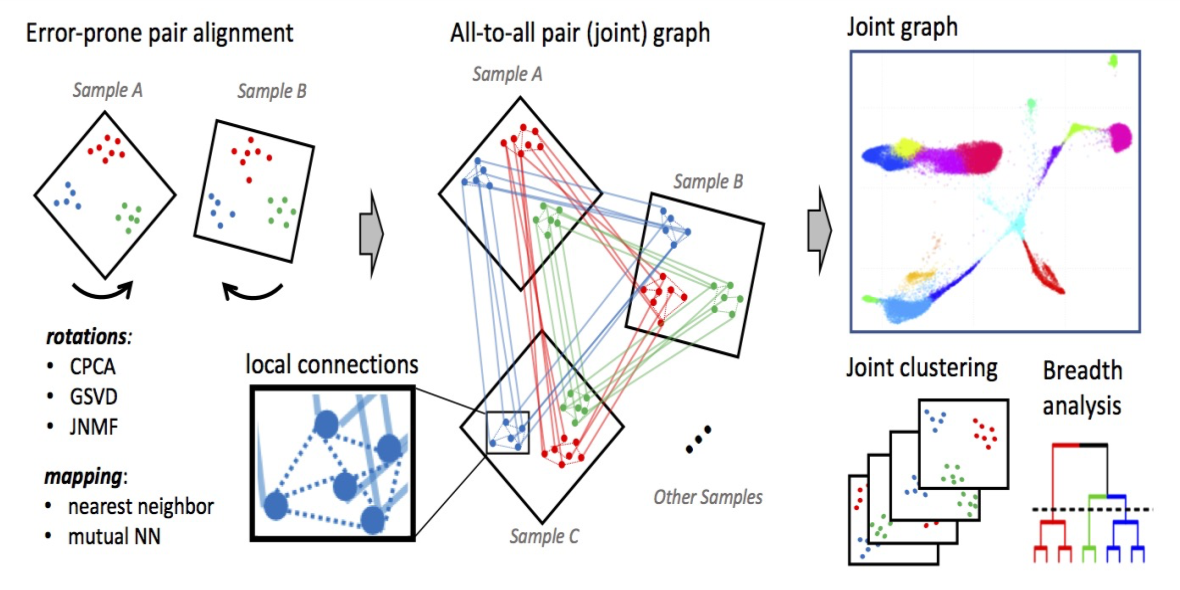
Figure16 | Conos原理示意图
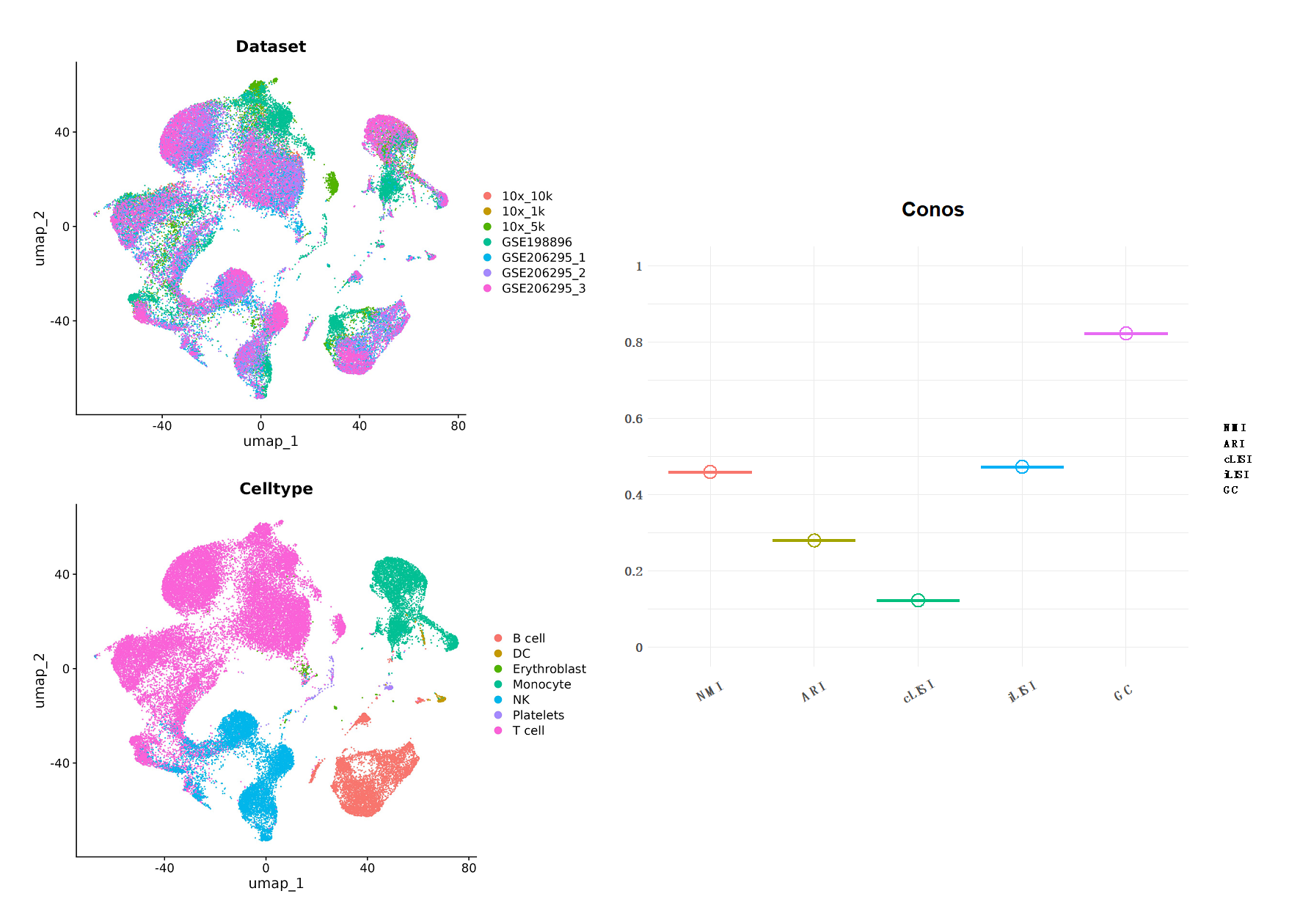
Figure17 | Conos 整合分析结果。左边为UMAP降维图形展示,分别以数据集和细胞类型分组;右图是结果评分
09 Scanorama
Scanorama[9]是另一个主要基于Python编写的工具。该方法的命名比较有趣,类似用于全景拼接的计算机视觉算法——识别具有重叠内容的图像并将其合并到更大的全景图(panorama) 中,Scanorama 可自动识别包含具有相似转录谱的细胞的scRNA-seq数据集,并利用这些匹配进行批次校正,实现数据的整合(Figure18)。首先使用一种改进的奇异值分解(SVD) 将原始基因表达数据转换为降维子空间,然后使用局部敏感哈希和随机投影森林方法执行最近邻搜索,以加快跨数据集间识别相互连接(即最近邻)的细胞。在我们的分析中,将其结合到Scanpy分析流程中,并选取结果中前30个主成分(此处指返回的Scanorama向量)进行后续分析。
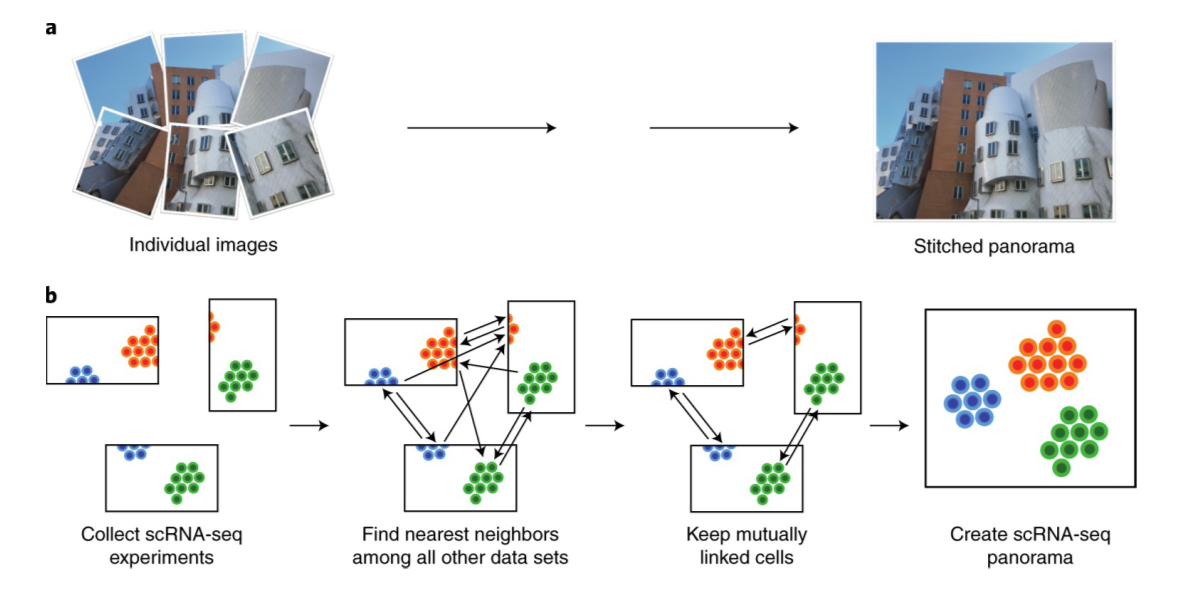
Figure18 | Scanorama 原理示意图。通过寻找最近邻来鉴定出共享细胞类型,所有匹配中互相连接的细胞被用于矫正批次效应,同时合并成一个数据集,这个过程形成了单细胞的全景图
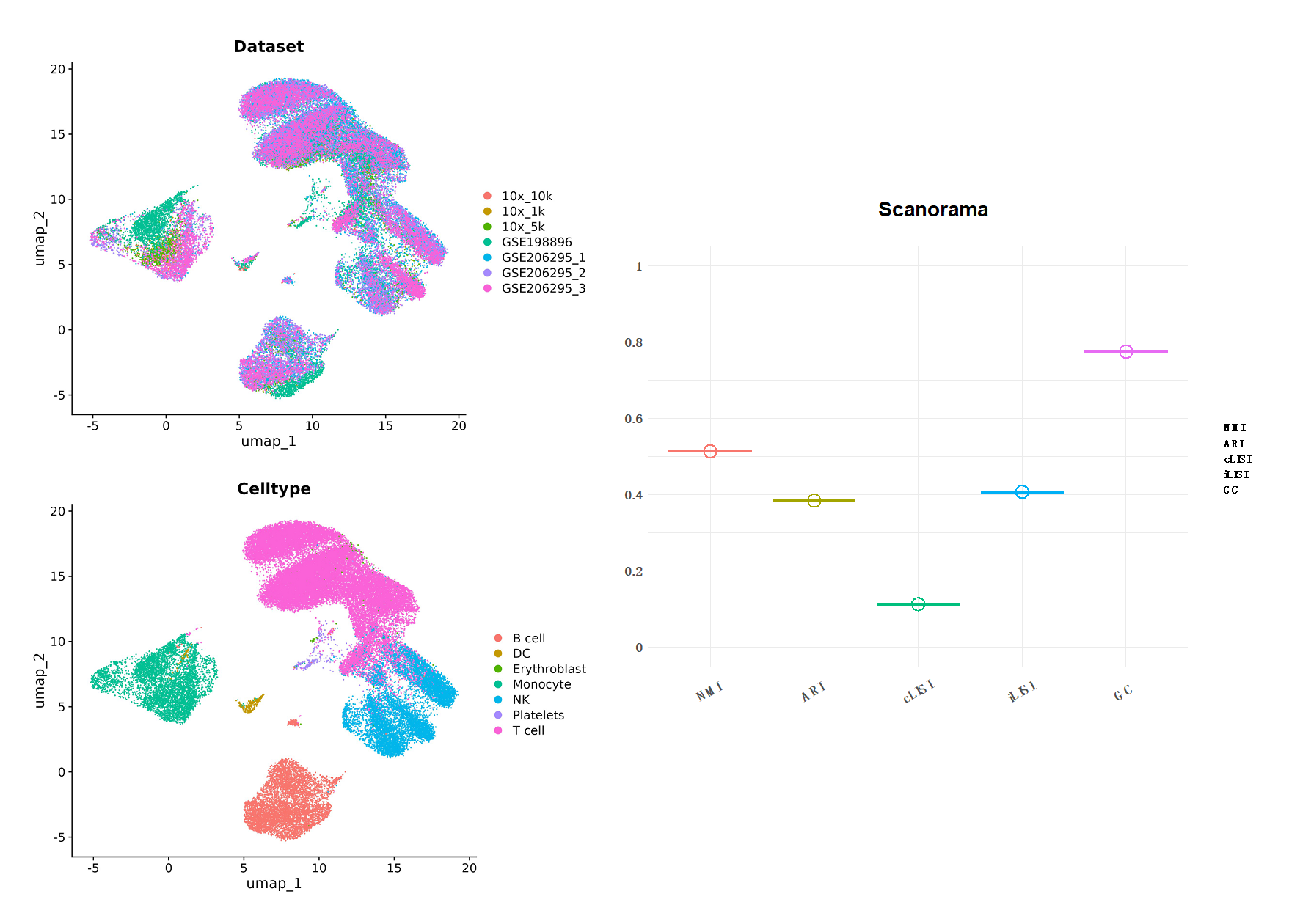
Figure19 | Scanorama 整合分析结果。左边为UMAP降维图形展示,分别以数据集和细胞类型分组;右图是结果评分
Conclusion
我们选取了7例PBMC样本(表1),预处理后共剩余46367个细胞。在直接合并分析的情况下,我们可以看见明显的批次效应(Figure2,3),虽然样本类型相同,但不同来源的数据集之间分散较为明显。为了能够综合评价结果,我们还采用了一些指标来对结果进行简单的打分,包括NMI、ARI、LISI和GC(表2),其中,LISI可以被用于衡量局部细胞类型分布(cLISI) 和局部批次分布(iLISI)。此外,这些统计指标可进一步分为两组,分别用于反映生物学效应的保留(NMI、ARI、cLISI)和批次效应的去除(GC、iLISI)。除了部分工具如Scanorama、ComBat是在Python环境下完成,大部分时间我们主要是在R环境下运行整合任务及绘图,而对于需要在Python中执行的任务,我们最后也会将其重新导出至R进行打分和图形绘制。
我们采用了常见的一共9种方法进行数据的整合。首先,从降维图形来看,这些不同的方法都很好地混合了不同批次来源的、相同类型的细胞(Figure4-20)。但是,部分方法存在混合不均匀的现象,如ComBat(Figure15),不同数据集来源的细胞仍然比较散乱,虽然大部分的细胞都按照相同类型聚集在一起,但是局部还是会有不同类型细胞重叠的情况;BBKNN虽然在数据集混合上表现一般,但相比ComBat之下,细胞类型分离较为清楚,这一点在cLISI分数上也有体现。从评分结果看(Figure21),在本次运行任务下,综合评分由高到低排序依次为Scanorama、Conos、Harmony、LIGER、SeuratCCA、fastMNN、SeuratRPCA、BBKNN和ComBat;而将评分拆分来看,LIGER、SeuratCCA和Harmony在去批次上表现更佳,Scanorama和Conos可能更倾向于保留生物学特征。由于单细胞数据的特性,类似ComBat这种开发用于bulkRNA测序、线性矫正方法可能不太适用于scRNA数据,这一点在很多文章中也有所提及[4];对于其它方法,虽然他们的基本原则相同,但是所采用的方法各异,例如fastMNN和SeuratCCA虽然都是在低纬嵌入上寻找MNNs,但所使用到的空间却有所不同,前者是利用主成分,后者则
选择了CCA方法,与此对比,BBKNN则是直接在近邻图上进行矫正。评分的结果提供给给我们量化方法好坏的可能性,但同时我们也不能忽略真实的生物学情况。对于一组未知细胞类型的样本来说,当某个方法错误估计了批次效应或强行合并了不匹配的细胞,很有可能造成评分分值很高但却存在过矫正现象的结果。因此,当我们看到整合结果中存在明显离群细胞,也不排除是由于独特的生物学差异所导致。额外需要注意的是,本次分析所选择的应用场景为——样本类型相同,批次不同。虽然比较单一,但使用较为广泛。在许多研究中我们可以看到,不同的评估方法结果差别很大,即使在相同的应用场景下,不同文章得出的结论也大相径庭[9-11]。这也提示我们,并没有一种方法可以真正做到 "One size fits all",我们需要按照实际情况做出相应的调整,以便更好地进行分析。
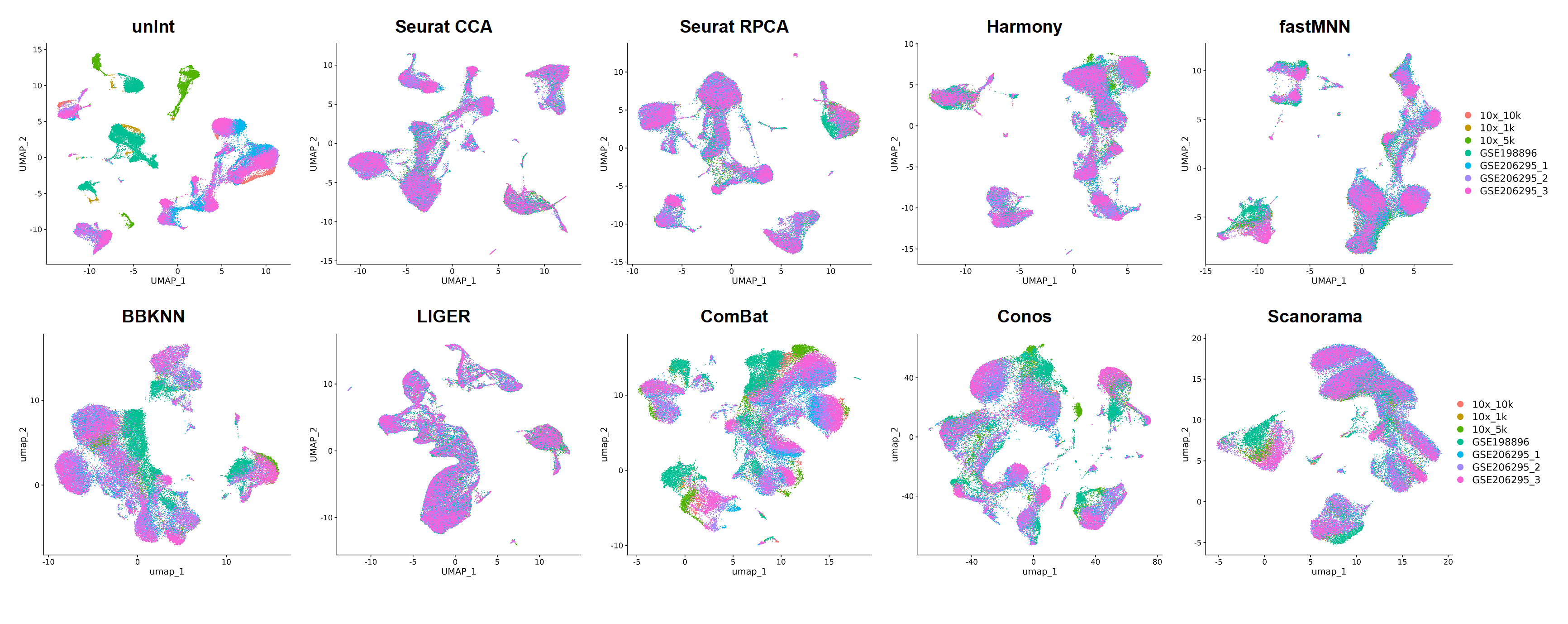
Figure20 | UMAP图展示不同方法整合结果,以样本来源进行分组
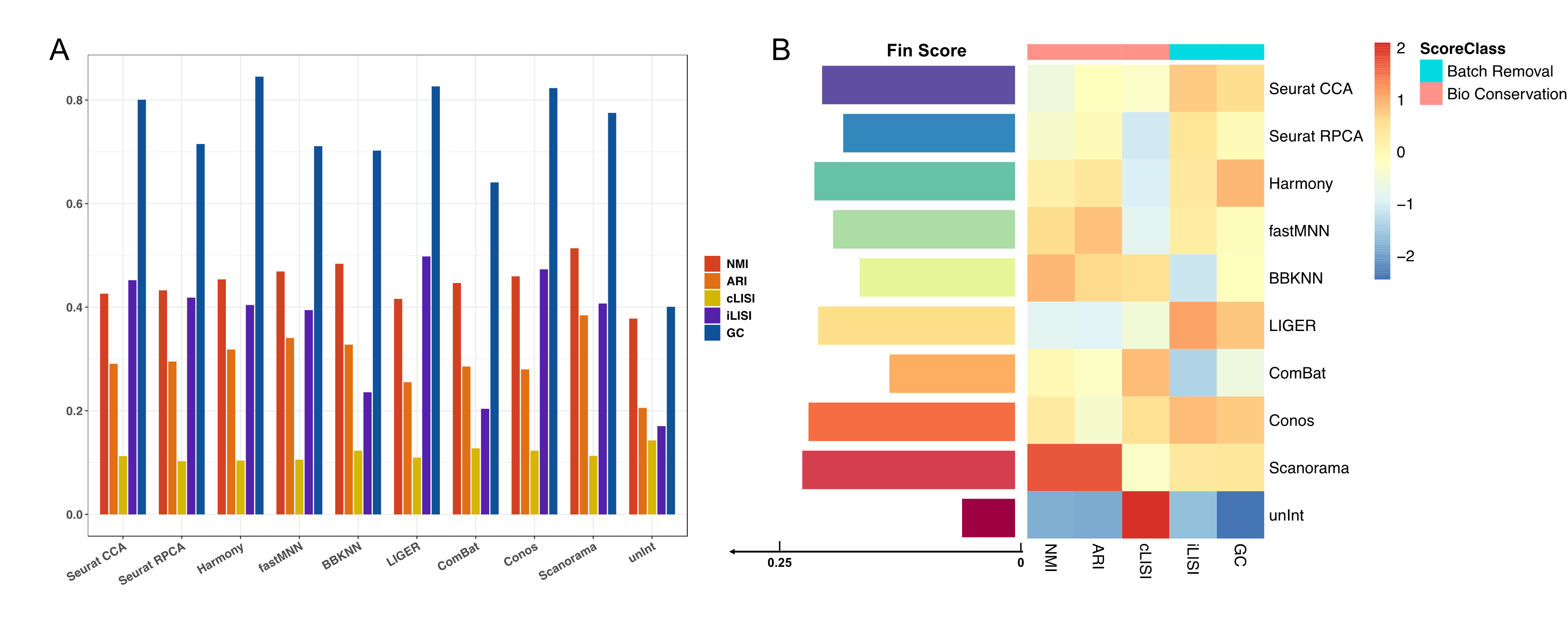
Figure21 | 不同方法各项评分展示。图A:柱形图分组表示各种方法在各项评估指标的具体分数值;图B:热图形式展示评分,左边为总计得分
表1:人外周血单个核细胞样本数据来源

表2:评分指标

References
[1] Aran, D., Looney, A.P., Liu, L. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 20, 163–172 (2019).
[2] Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019 Jun 13;177(7):1888-1902.e21.
[3] Korsunsky, I., Millard, N., Fan, J. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296 (2019).
[4] Haghverdi, L., Lun, A., Morgan, M. et al. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol 36, 421–427 (2018).
[5] Polański K, Young MD, Miao Z, Meyer KB, Teichmann SA, Park JE. BBKNN: fast batch alignment of single cell transcriptomes. Bioinformatics. 2020 Feb 1;36(3):964-965.
[6]Welch JD, Kozareva V, Ferreira A, Vanderburg C, Martin C, Macosko EZ. Single-Cell Multi-omic Integration Compares and Contrasts Features of Brain Cell Identity. Cell. 2019 Jun 13;177(7):1873-1887.e17.
[7] Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007 Jan;8(1):118-27.
[8] Barkas, N., Petukhov, V., Nikolaeva, D. et al. Joint analysis of heterogeneous single-cell RNA-seq dataset collections. Nat Methods 16, 695–698 (2019).
[9] Hie, B., Bryson, B. & Berger, B. Efficient integration of heterogeneous single-cell transcriptomes using Scanorama. Nat Biotechnol 37, 685–691 (2019).
[10] Luecken, M.D., Büttner, M., Chaichoompu, K. et al. Benchmarking atlas-level data integration in single-cell genomics. Nat Methods 19,
[11] Tran, H.T.N., Ang, K.S., Chevrier, M. et al. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol 21, 12 (2020).
[12] Argelaguet, R., Cuomo, A.S.E., Stegle, O. et al. Computational principles and challenges in single-cell data integration. Nat Biotechnol 39, 1202–1215 (2021).