- 移动端


菁良科技(深圳)有限公司
4 年
手机商铺
- NaN
- 0.2999999999999998
- 1.2999999999999998
- 0.2999999999999998
- 3.3
推荐产品


公司新闻/正文
原则解读 | 地中海贫血相关基因检测试剂注册技术审查指导原则
1568 人阅读发布时间:2020-03-20 11:48
近期国家药品监督管理局医疗器械技术审评中心发布了地中海贫血相关基因检测试剂注册技术审查指导原则,该指导原则旨在指导注册申请人对地中海贫血相关基因检测试剂注册申报资料的准备及撰写,同时也为技术审评部门对注册申报资料的技术审评提供参考。
该指导原则的技术要求是基于PCR探针法方法建立的,并适用于进行首次注册申报和相关许可事项变更的产品。
小编主要就企业参考盘和关于性能评价该指导原则的要求和跟大家分享一下。
企业参考盘要求
企业参考品
企业参考品主要包括阳性参考品、阴性参考品、检测限参考品和精密度参考品等。申请人应提交企业参考品的原料选择、制备方法、基因序列确认及检验标准的详细研究资料等。
- 阳性参考品
可采用临床样本或其核酸溶液,如采用临床样本,则样本类型应与待测样本一致。应至少包含所有检测位点的杂合型样本,同时尽量纳入纯合突变型样本。对于某些稀有基因型,也可采用细胞系或质粒。
- 阴性参考品
- 检测限参考品
- 精密度参考品
目前,该类产品已有国家参考品,对于申报产品所声称的检测位点,企业参考品的要求应不低于国家参考品。
性能评价要求
分析性能评估资料
申请人应针对下述各项分析性能提交详细的评估资料,包括试验地点、适用仪器、试剂规格、批号、试验方法、试验样本(类型、来源、数量、处理方法、基因型和浓度确认等)、可接受标准、统计方法、试验数据及结论等。分析性能评估的实验方法可以参考国内外有关体外诊断产品性能评估的指导原则。
每项性能评估应尽量采用与适用样本类型一致的临床样本,对于某些稀有基因型,也可采用细胞系等。
- 核酸提取/纯化性能(如有)
- 检测准确性
- 分析特异性
- 应针对同源序列或检测范围外基因和位点进行交叉反应验证,说明交叉反应样本的制备方法、核酸序列确认方法,提交详细的验证资料。
- 应针对可能的内源和外源性干扰物进行干扰试验研究。内源干扰物主要涉及血脂、胆红素、血红蛋白和白蛋白、母体细胞的干扰等,外源干扰物主要包括血液样本采集可能用到的抗凝剂和常用药物等。
干扰试验可通过在临床样本中人工添加干扰物质的方式,评价干扰物质对目标序列检测的影响,也可直接采集暴露于干扰因素后的受试者样本,进行干扰试验评价。建议申请人在每种干扰物质的潜在最大浓度(“最差条件”)条件下进行评价;如有干扰,应确定不产生干扰的最高浓度。
- 精密度评价
精密度评价应采用临床样本进行试验,试验操作完全按照说明书执行,包含核酸提取/纯化等步骤(如有)。应对每一反应体系进行精密度评价,并至少覆盖代表性的突变位点和突变类型。
精密度评价需满足如下要求:
- 对可能影响检测精密度的主要因素进行验证,除检测试剂本身外,还包括分析仪器、操作者、地点、时间、检测轮次和试剂批次等。
- 设定合理的精密度评价周期,对批内/批间、日内/日间以及不同操作者之间的精密度进行综合评价。如有条件,申请人应选择不同的实验室进行重复实验以对室间重复性进行评价。
- 用于精密度评价的临床样本至少设置低浓度和中/高浓度水平。
- 精密度指标可设置为CV等(如有),申请人应对精密度指标评价标准做出合理要求。
阳性判断值确定资料
建议纳入一定数量的临床样本,应包括所有检测位点的各种基因型(野生型、杂合突变型,同时尽量纳入纯合突变型),采用受试者工作特征(ROC)曲线或其他合理方法确定阳性判断值。
如试剂判读存在灰区,应解释说明灰区范围的确定方法。
对于某些检测方法学,阳性判断值研究可能不适用,申请人应说明理由。
稳定性研究资料
稳定性研究资料主要包括申报产品的稳定性研究和适用样本的稳定性研究两部分。前者主要包括申报产品的实时稳定性、开瓶/复溶稳定性、运输稳定性及冻融次数限制的研究等;后者则是指适用样本的保存条件和保存时间等的研究。
实时稳定性研究应采用至少三批样品在实际储存条件下保存至成品有效期后,选取多个时间点进行产品性能评价,从而确定产品保存条件和有效期。
如核酸提取液可保存,还需对核酸提取液的保存条件和保存时间进行研究。
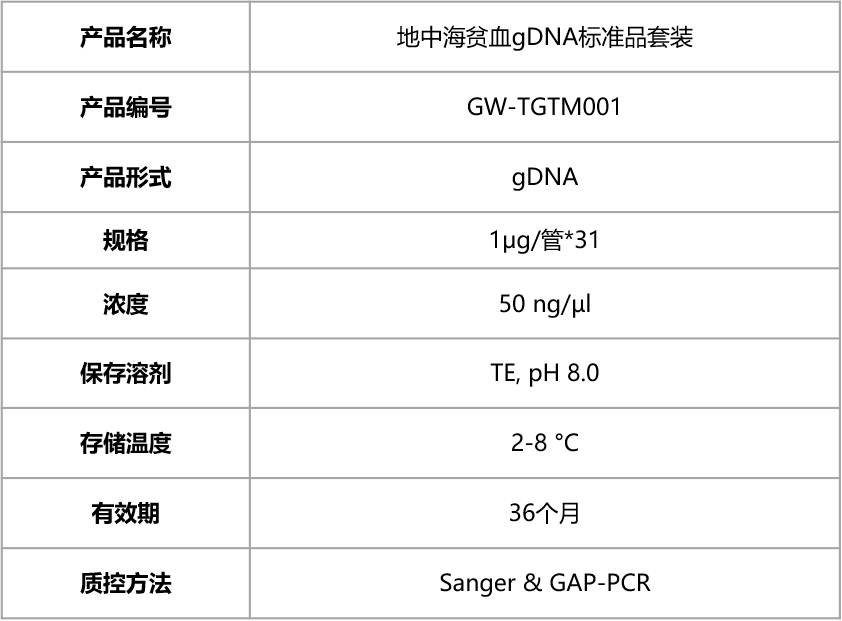
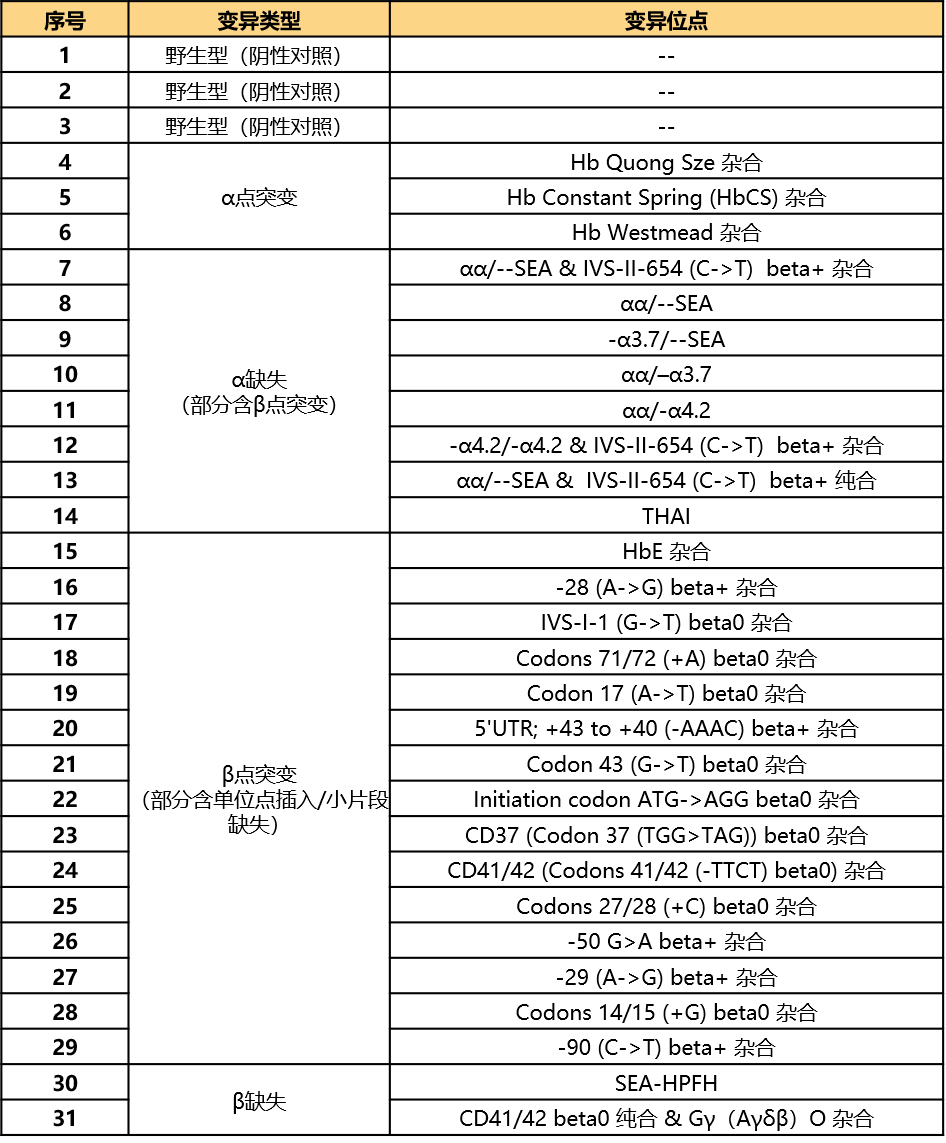
菁良地中海贫血gDNA标准品套装
菁良基因地中海贫血gDNA标准品套装包含常见的α,β地中海贫血突变,可用于地中海贫血基因检测试剂盒的注册报证及阴阳性质控品,以及检测流程的日常质控与性能评价。
产品特点
◆ 样本来源于临床样本永生化细胞系
最大程度接近临床样本并稳定可重复
◆ 变异位点全
覆盖国家参考品所有变异位点
◆ 适用范围广
PCR及NGS等平台皆适用
◆ 多种平台验证变异位点
Sanger 测序--点突变
GAP-PCR--片段缺失
◆ 阴性及阳性样本配套
套装形式(31管):28管阳性样本+3管阴性样本
◆ 样品稳定,批间差异小
产品参数