

产品分类
公司荣誉

公司图片












联系我们
- 公司地址
-
上海市闵行区联航路1239号8B楼3层
邮编:200233
- 联系电话
- 021-****6157登录查看商家电话
- 传真号码
- 021-34786153
- 电子邮箱
- gulingzhi@****bio.com登录查看商家邮箱
- 公司网址
- www.yingbio.com

最新研究动态
胃癌中索拉菲尼诱导铁死亡实锤——ATF2
发布时间:2024-03-07 08:52 | 点击次数:
索拉非尼是一种酪氨酸激酶抑制剂,在包括胃癌(GC)在内的多种癌症中作为铁死亡诱导剂具有重要的抗肿瘤作用。然而,索拉非尼作为铁死亡诱导剂的作用最近受到了质疑。有关铁死亡与ATF2关系的信息非常有限,而ATF2在索拉非尼诱导的铁死亡中的作用也尚未研究。因而,本研究探讨了GC中ATF2在索拉非尼诱导的铁死亡中的作用及其分子机制。作者发现索拉非尼激活ATF2的表达,并进一步促进HSPH1的表达,减少SLC7A11蛋白的降解,从而导致对脂质过氧化的保护。在小鼠中,敲除ATF2能有效提高索拉非尼的敏感性。本文于2023年2月发表在《Redox Biology》IF: 11.4期刊上。
技术路线

主要实验结果
1、ATF2在胃癌中表达上调,预示预后不良
TCGA数据显示ATF2在胃癌组织中的高表达(图1A),ATF2在GC细胞系中的表达也显著高于非恶性细胞系(图1B),在12对临床样本中,ATF2在胃癌组织中的表达也显著高于癌旁(图1C)。在107例GC组织和22例癌旁组织的GC芯片中检测了ATF2的表达(图1D),61.7%的GC组织高表达ATF2(图1E),并且ATF2高表达的GC患者生存期更短(图1F)。以上表明ATF2在胃癌中表达上调,预示预后不良。

图1 ATF2在胃癌中表达上调,预示预后不良
2、ATF2敲除抑制GC细胞恶性表型
如图2所示,过表达ATF2促进细胞增殖、迁移和侵袭,敲低ATF2则结果相反。表明ATF2促进GC恶性表型。
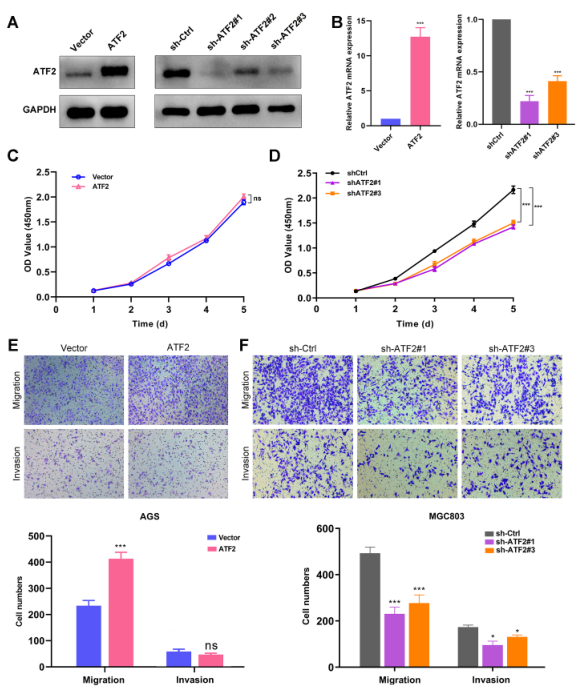
图2 ATF2敲除抑制GC细胞恶性表型
3、索拉非尼诱导GC细胞铁死亡并激活ATF2表达
由于先前的研究表明索拉非尼诱导的铁死亡中存在不确定性,所以作者首先研究了索拉非尼是否会诱导GC细胞的铁死亡。如图3A所示,10 μM索拉非尼作用24 h后,AGS和MGC803细胞形态均缩小、变圆、排列疏松,而与铁死亡抑制剂共同处理可以部分逆转这些变化。与对照组相比,索拉非尼治疗导致总细胞ROS和脂质ROS增加(图3B-3C)。此外,用索拉非尼孵育导致MDA急剧增加而GSH下降,而这种影响被fer-1有效抑制(图3D和E)。以上结果表明索拉非尼诱导GC细胞铁死亡。

图3索拉非尼诱导AGS和MGC-803细胞铁死亡
鉴于铁死亡与线粒体功能密切相关,接下来检测线粒体膜电位(MMP)和形态学的变化。JC-1染色结果显示,与对照组相比,索拉非尼治疗显著降低MMP(图4A),透射电镜显示,索拉非尼处理的MGC803细胞线粒体嵴明显减少或缺失,线粒体膜密度增加(图4B)。这些结果也表明索拉非尼可以诱导GC细胞的铁死亡。作为一种关键的应激反应转录因子,ATF2在索拉非尼诱导的铁死亡反应中的表达变化仍然未知。如图4C所示,索拉非尼处理增加ATF2的表达,尤其是磷酸化形式。值得注意的是,免疫荧光显示,索拉非尼刺激后 ATF2 在细胞核中的表达量更高(图4D)。因此,索拉非尼诱导的铁变态反应可能会促进 ATF2 的核转位并增强 ATF2 在 GC 细胞中的转录活性。
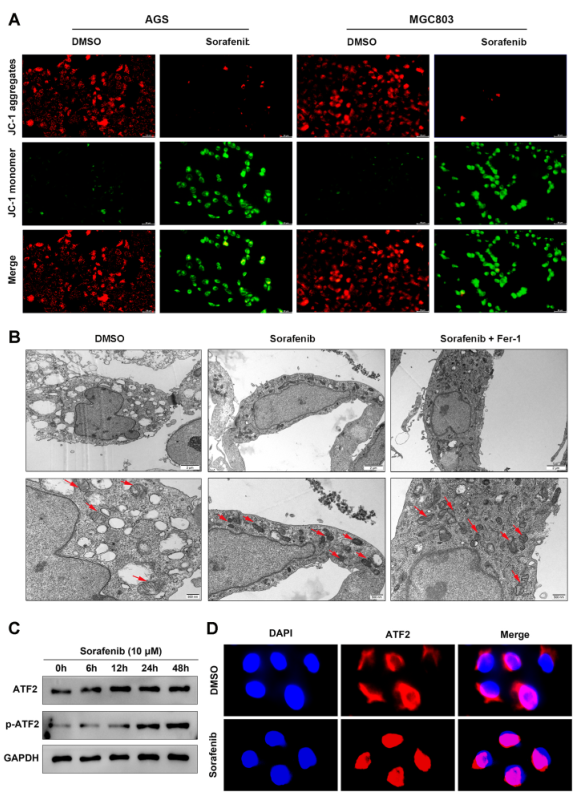
图4索拉非尼治疗增加GC细胞中ATF2的表达并促进其核易位
4、ATF2敲除抑制索拉非尼诱导的GC细胞铁死亡
如图5所示,为了解ATF2在索拉非尼诱导的铁死亡中的作用,作者干预ATF2的表达水平并评估它们对铁死亡的影响。与载体组相比,ATF2过表达导致AGS细胞中索拉非尼的IC50值升高,而ATF2敲低导致MGC803细胞中索拉非尼的IC50值降低(图5A)。索拉非尼治疗和ATF2敲低均升高了细胞总ROS和脂质ROS,ATF2敲低导致AGS细胞中ROS的进一步升高,而ATF2过表达抑制了索拉非尼在MGC803细胞中诱导的升高(图5B-C)。在索拉非尼处理的GC细胞中,ATF2过表达导致MDA降低,GSH升高,而ATF2敲低则显示相反的结果(图5D-E)。TEM结果显示索拉非尼治疗后,ATF2敲低的GC细胞线粒体减少,膜密度增加,嵴退化,这种作用部分被ATF2过表达逆转(图5F)。这些发现表明ATF2过表达抑制索拉非尼诱导的GC细胞铁死亡。
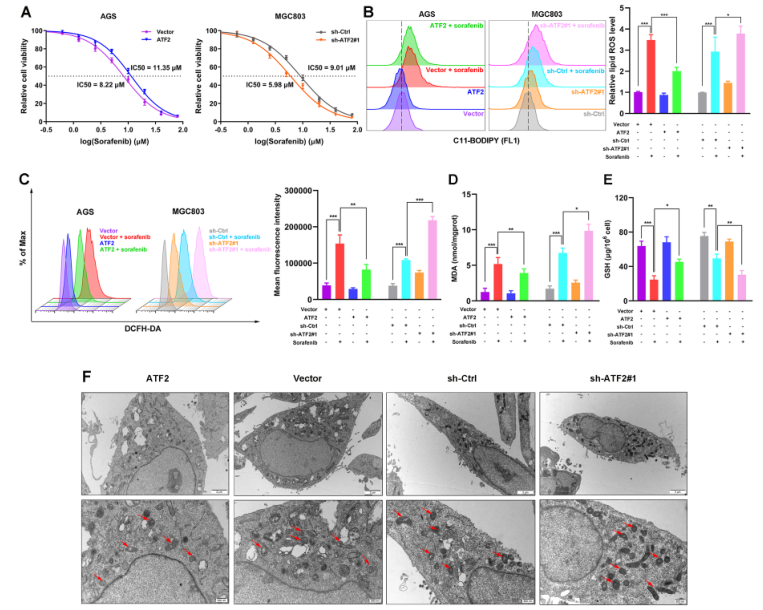
图5 ATF2敲除抑制索拉非尼诱导的GC细胞铁死亡
5、ATF2在GC细胞中激活HSPH1的转录
为进一步阐明其潜在机制,进行RNA-seq和ChIP-seq分析,以确定全基因组的DNA结合位点和ATF2的潜在转录靶点。如图6A所示,RNA-seq分析显示,与对照组相比,MGC803细胞中ATF2基因敲除后,有1059个基因上调,370个基因下调。重要的是,RNA-seq数据的通路富集分析表明,ATF2基因敲除会显著影响铁死亡通路(图6B)。接下来,ChIP-seq共鉴定出24119个峰,对应3641个RefSeq基因,其中2.88%位于启动子-转录起始位点(图6C)。在染色体上观察到了不同的峰值,并扫描了峰值之间共享的motif(图6D和E)。
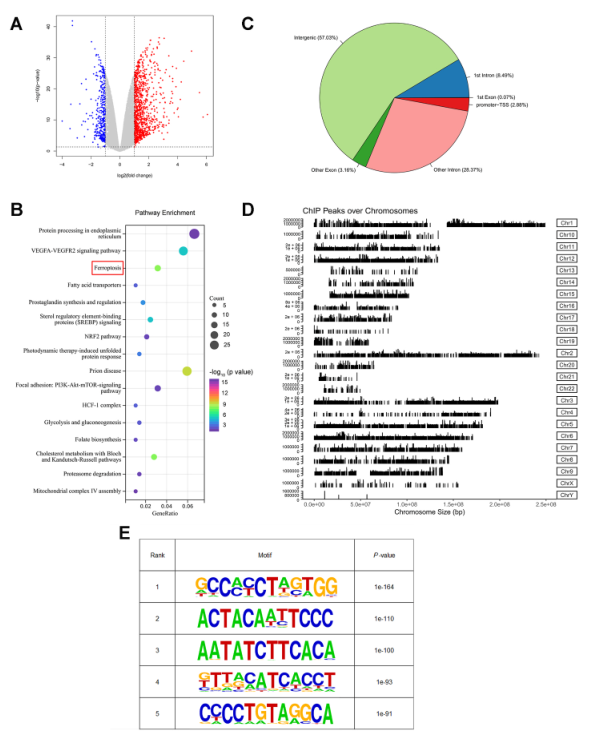
图6利用RNA-seq和ChIP-seq技术鉴定ATF2的全基因组DNA结合位点和转录靶点
然后,对RNA-seq和ChIP-seq数据进行交叉分析,筛选出222个ATF2直接调控的候选转录靶点(图7A)。其中,HSPH1在ATF2敲低后显著下调(图7B)。此外,在TCGA数据集中,HSPH1在GC中的表达与ATF2呈显著正相关(图7C)。如图7D所示,ChIP-seq数据显示HSPH1启动子区域存在显著的ATF2结合峰。因此,作者推测HSPH1可能是ATF2的潜在靶基因。WB分析结果显示,ATF2过表达后,HSPH1增加,而ATF2敲除后,HSPH1减少(图7E)。此外,ChIP-qPCR进一步证实ATF2可与HSPH1的启动子区域结合(图7F)。这些数据表明ATF2可以通过与HSPH1启动子结合来激活HSPH1的表达。
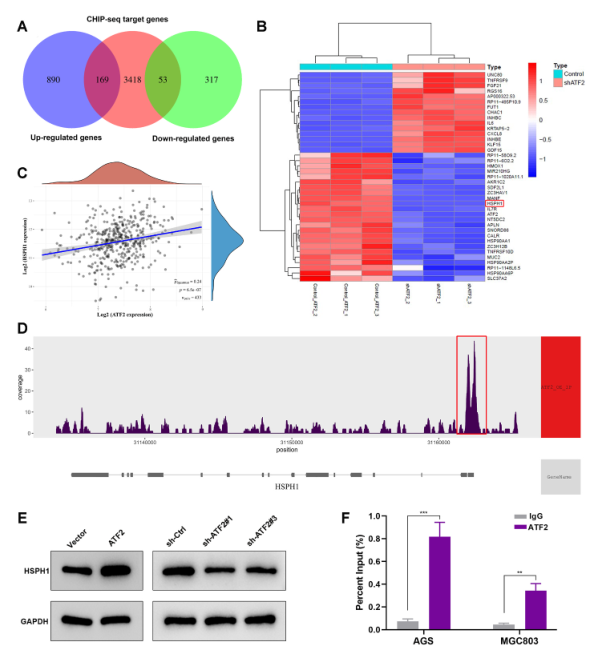
图7 ATF2结合HSPH1启动子激活其转录
6、HSPH1与SLC7A11在GC中相互作用并增加其稳定性
鉴于多项研究报道了热休克蛋白在铁死亡反应中的突出作用,作者试图确定 HSPH1是否会影响SLC7A11在GC中的表达。首先查询GeneMANIA数据库,预测并显示HSPH1与SLC7A11之间潜在的相互作用(图8A)。为验证这一预测,进行co-IP实验,确定HSPH1与SLC7A11存在物理相互作用(图8B)。采用siRNA敲除HSPH1的表达(图8C),并将HSPH1 siRNA转染到ATF2稳定过表达的AGS细胞中。有趣的是,观察到HSPH1的敲除降低了SLC7A11的蛋白表达水平,但没有降低mRNA水平(图8D)。因此,进行CHX追逐试验,以确定在是否敲除HSPH1的情况下SLC7A11蛋白的半衰期。WB分析表明,与对照组相比,抑制HSPH1会加速AGS细胞中SLC7A11蛋白的降解(图8E)。这些结果表明,HSPH1可以与SLC7A11相互作用,并至少部分地通过增加SLC7A11蛋白的稳定性来增加其表达。敲除HSPH1可部分消除ATF2过表达对细胞ROS和脂质ROS的影响(图8F和G)。同样,联合转染HSPH1 siRNA能显著逆转ATF2过表达对铁变态细胞死亡中细胞MDA和GSH水平的影响(图8H和I)。这些结果为ATF2通过HSPH1调控索拉非尼诱导的GC细胞铁死亡提供了证据。
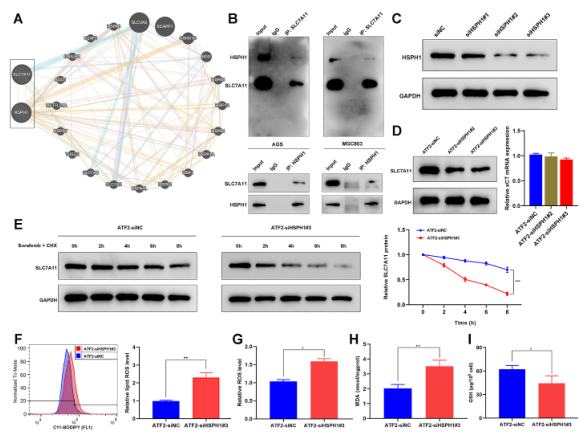
图8 HSPH1与SLC7A11相互作用,提高其在GC中的稳定性
7、体内ATF2敲低增加GC对索拉非尼的敏感性
最后,为评估ATF2单独和联合索拉非尼对体内GC的影响,建立裸鼠异种移植模型(图9A)。稳定ATF2敲除细胞形成的肿瘤始终比对照GC细胞形成的肿瘤更小更轻,这表明ATF2敲除能有效抑制体内肿瘤的生长(图9B)。此外,经索拉非尼治疗后,皮下肿瘤的体积和重量均显著减少(图9C和D)。也就是说,ATF2敲除联合索拉非尼治疗对肿瘤生长的抑制作用最强。为更好地观察潜在的死亡,皮下肿瘤切片被4-HNE染色,IHC染色显示,sh-ATF2 +索拉非尼组的4-HNE 表达量最高(图9E)。因此,ATF2基因敲除可增强索拉非尼在体内对GC的抗肿瘤作用。
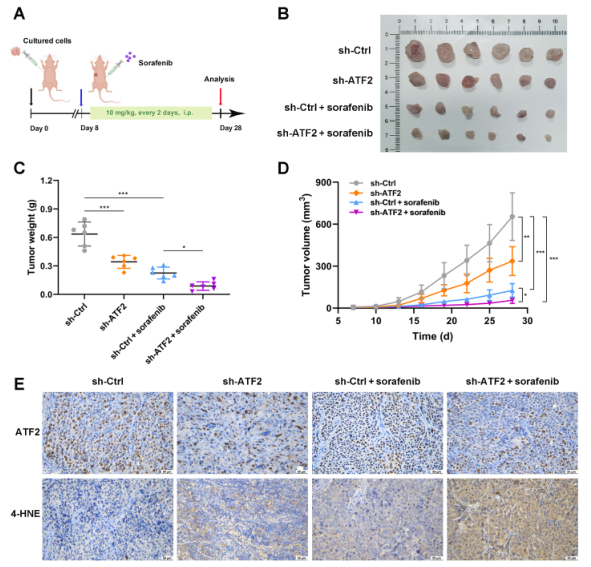
图9体内ATF2敲低增加GC对索拉非尼的敏感性
总之,在本研究中,作者发现索拉非尼激活ATF2的表达,并进一步促进HSPH1的表达,减少SLC7A11蛋白的降解,从而导致对脂质过氧化的保护(图10)。因此,以ATF2/HSPH1轴为靶点来增强索拉非尼诱导的铁死亡代表了一种有吸引力的GC治疗策略。

图10 图像摘要
实验方法
胃癌临床样本采集,细胞培养和转染,WB,RT‒PCR,IHC,细胞生长曲线,IC50检测,Transwell迁移和侵袭,Calcein-AM/PI染色,ROS和脂质过氧化检测,MDA和GSH检测,线粒体膜电位检测,透射电镜TEM,RNA-seq,ChIP-seq,共免疫沉淀(Co-IP),蛋白稳定性CHX实验,小鼠模型,
参考文献
Xu X, Li Y, Wu Y, Wang M, Lu Y, Fang Z, Wang H, Li Y. Increased ATF2 expression predicts poor prognosis and inhibits sorafenib-induced ferroptosis in gastric cancer. Redox Biol. 2023 Feb;59:102564. doi: 10.1016/j.redox.2022.102564. Epub 2022 Dec 2. PMID: 36473315; PMCID: PMC9723522.