大家都在搜

| 目的基因在细胞水平上的导入方式可以通过瞬转的方式进行,但想要长期在目的细胞中研究基因功能,需要建立稳转细胞系,可以降低频繁转染或者病毒包装的成本,方便实验研究。接下来,小编将对整个筛选流程进行一个简单的介绍。 一、慢病毒介导的稳转株筛选原理
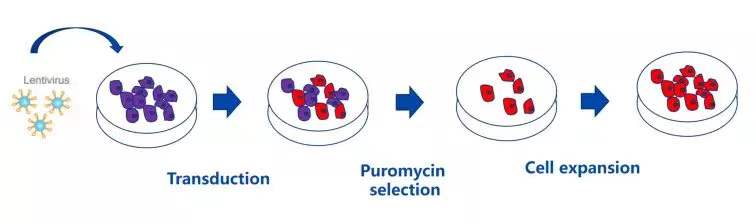
筛选流程示意图
实验原理:通过将外源 DNA/shRNA 克隆到带有某种抗性基因的载体上,重组载体转染至宿主细胞并整合到其染色体中,并随细胞分裂稳定传递,最后用载体中所含抗性基因进行筛选。
抗性选择:常用的真核表达载体抗性筛选标志物有嘌呤霉素 (puromycin)、新霉素 (neomycin) 和杀稻瘟菌素(blasticidin)。 二、准备及预实验-MOI 摸索及抗性浓度摸索 慢病毒感染 MOI 预实验 根据《慢病毒感染手册》筛选出该细胞的 MOI 值。
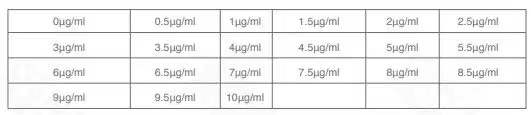
Puromycin 工作浓度筛选预实验 大部分细胞 puromycin 的工作浓度为 1~10 μg/ml 在《慢病毒感染手册》上查阅 Puromycin 目的细胞中筛选的致死筛选参考用量。部分细胞的参考用量可参见附录 2《常见细胞 MOI 及感染条件》,请在筛选时设置未感染病毒的野生型细胞对照,加入等量浓度的 Puromycin。 如未在附件中找到使用的细胞,在正式实验前,先进行 Puromycin 梯度筛选预实验(确定能在 2 天杀死野生型细胞的最低 Puromycin 浓度)。具体步骤如下: 1. 提前 24 h 将细胞铺入 24 孔板,细胞量根据细胞生长状态及大小调节,在 24 h 后,细胞密度在 70% 左右; 2. 细胞生长 24 h 后,加入不同终浓度的 puromycin,如图所示;
Puromycin 浓度分组情况
3. 再培养 48 h,显微镜下观察,将全部杀死细胞的最低浓度的 puro 组作为工作浓度。 三、稳定株筛选 慢病毒感染 48-72 h 后(70-80% 融合度),将细胞继续培养于含适当浓度 Puromycin 的培养液中,筛选 48 h 后,空细胞加 puro 组全部死亡,我们认为感染病毒组剩下全部是阳性细胞,进行以下操作:
A. 混合克隆稳转株的构建
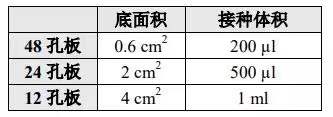
1. 目的细胞病毒感染 处于对数生长期的细胞胰酶消化,完全培养基制成 3-5×104 个/mL 细胞悬液,并根据表 1 接种相应的细胞数到培养板中,继续培养保证感染时铺板量达到 15-30% 左右,按照预实验结果,参考表 1 更换感染培养基,加入最适病毒量进行感染。参照预实验结果,选择感染后最适时间点更换为常规培养基继续培养,一般为感染后 8-16 h 左右。
按照预实验确定的感染条件,使用感染液将细胞制成 3-5×105 个/mL 悬液,并根据表 2 接种相应的细胞数到培养板中。铺板后无需培养,参照预实验确定的 MOI 加入最适病毒量感染。参照预实验结果,选择感染后最适时间点,一般为 8-16 h 左右,将各孔中细胞收集到干净的 1.5 ml EP 管中,以 1300 rpm 离心 2 min,去掉上清液,更换为完全培养基,轻轻混匀后放回培养板中继续培养。
2. 感染后细胞加入抗生素筛选 1)感染 72 h 后,观察细胞状态和感染效率。要求细胞状态良好,未出现大量死亡现象,特别是保证 NC 组与 CON 组细胞状态相当。 2)加入合适浓度的抗生素,筛选至少 48 h。如病毒载体带有荧光标记,荧光效率需要达到 100% 后,降低抗生素维持浓度(相较之前的药筛浓度,浓度减至之前浓度的 1/2~1/4 或更低)继续对感染后的细胞进行筛选和扩增,同时收集细胞进行下游 qPCR 检测(鉴定目的基因表达水平)。
3. 细胞扩增、冻存、复检 1)将加入抗生素维持浓度的细胞继续进行扩增。待 qPCR 检测结果合格后进行冻存,保证每株细胞至少冻存 6 支。 2)冻存 2-3 天后,任意复苏一支,判断复苏细胞状态。
B. 单克隆稳转株的构建
对感染并筛选后的混合克隆稳转株细胞进行稀释培养,挑取单一细胞生长而成的细胞克隆,再进行扩大培养,以获得性状单一、表达稳定的细胞株: 具体步骤如下: 1)将细胞消化后接种于 96 孔板中,接种的细胞密度为 1 个/孔(可通过有限稀释的方法,也可通过流式分选); 2)标记出具有单个细胞的孔等细胞扩增后用 puro 进行筛选; 3)筛选完毕后,收集细胞进行 qPCR 或 Western Blot 鉴定,选择鉴定结果正常的单克隆细胞冻存保种
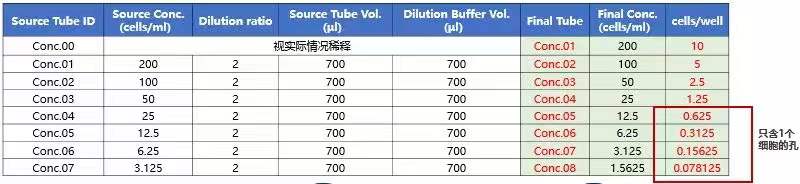
1)倍比稀释筛选 细胞计数设定首排的浓度,例如首孔设置 10* cells/孔,每孔 50*μl(细胞浓度:100* cells/ml),96 孔板补足最终培养体积 100ul。
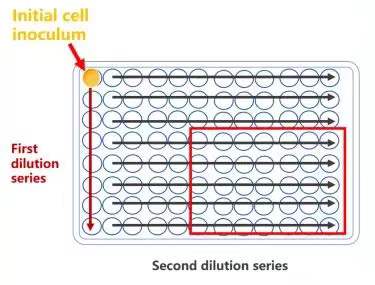
2)两步系列稀释筛选
稀释倍数参考:以首孔浓度选择 1E4cell/孔 * 举例,标红区域出现 1 个细胞的概率更大 3)直接稀释至 1 个/孔:以 96 孔板为例,将细胞消化后接种于 96 孔板中,接种的细胞密度为 1 个/孔。
前面介绍的都是关于单个病毒的稳转株构建方法,如果是两个病毒的话,具体应该如何操作呢?下面,小编以吉凯基因 CRISPR/Cas9 双载体慢病毒为例进行介绍。
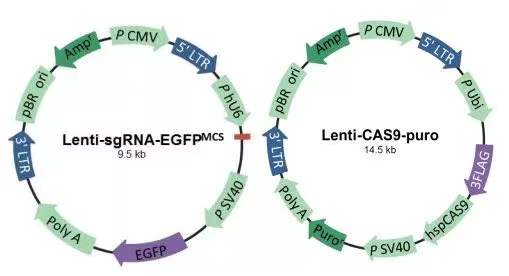
首先,让我们了解一下载体系统:
1. 双载系统:通过两个慢病毒分别向细胞导入 Cas9 蛋白和 sgRNA 序列表达框,从而实现对目的基因的敲除。Cas9 蛋白载体带有嘌呤霉素标记,sgRNA 带有绿色荧光标记/红色荧光/新霉素抗性可选。
2. 具体实验步骤: 1)Cas9 慢病毒稳转株构建 先感染 Cas9 慢病毒,参照《慢病毒感染手册》,选择 MOI 在 70-80% 的病毒剂量进行 Cas9 慢病毒稳转株的构建,经合适浓度的 Puromycin 抗药性筛选,得到稳定表达 Cas9 的混合克隆细胞株(待荧光为 100% 后将 puromycin 浓度减至之前浓度的 1/2~1/4 或更低维持); 2)Cas9 慢病毒稳转株的冻存 可以先将构建好的 Cas9 稳转株冻存作为工具细胞株,后续其他实验也可用该细胞株进行; 3)sgRNA 慢病毒感染 a. MOI 摸索预实验:可参考 Cas9 慢病毒 MOI 进行,也可以用 sgRNA 对照慢病毒进行预实验,摸索合适的 MOI,选择 MOI 在 70-80% 的病毒剂量进行 sgRNA 慢病毒感染实验; b. 筛选靶点有效性:分别使用 sgRNA 慢病毒对 Cas9 稳转株进行感染,判断靶点有效性,选取有效的 sgRNA 慢病毒进行后续稳转株的构建。 c. 有效 Cas9-sgRNA 稳转株的构建:筛选出有效靶点后,可进行多克隆、单克隆稳转株的构建(如需构建单克隆稳转株,建议筛选出有效靶点后再进行单克隆稳转株筛选,否则会导致工作量较大)。 d. 报告基因的选择及后续维持:一般第二次感染病毒的报告基因默认选择新霉素抗性,除此之外也可选择荧光标签进行筛选,需要注意的一点是,报告基因的选择的选择决定了后续稳转株的筛选方式(真核抗性筛选 or 流式分选)
与单载体系统相比,双载体系统需要两个病毒进行感染,同时可以得到 Cas9 慢病毒稳转株作为工具细胞株,大家可以根据自己的实验需求进行选择,把控好病毒的感染时间、调整好细胞状态,同时也要提前考虑好选择两种不同的抗性筛选方式,从而完成稳转株的筛选。 |

更有优质直播、研选好物、福利活动等你来!