大家都在搜

| 通过高通量测序等技术“读”DNA,让遗传信息解读和应用快速普惠大众,为“基因1.0时代”;正在酝酿的“基因2.0时代”,通过基因合成“写”DNA,深度理解、设计和重构生物系统,赋能育种、能源和环境等。预见未来,本文从技术角度一览基因合成原理和流程等。 划重点 1. 基因合成特点:设计、改造、验证、修正的反复试错 。 2. 核心点:基于计算机的DNA序列设计,成本、错误率和DNA序列转入细胞的功能测试。 3. 基因合成复杂度,从单链寡核苷酸链、双链寡核苷酸链、原核基因组到真核基因组。 4. 基因合成技术,包括基于DNA连接酶、基于DNA聚合酶两种思路。 5. 基因组完成组装后,需要依托基因组编辑技术进行验证并进行部分校正。
一 生命健康时代的宠儿:基因合成 BT(生物技术)和IT(信息技术)的结合,形成基因技术以超摩尔定律的速度普及,在医疗、健康、大数据和金融层面逐渐形成共识,工业时代的技术革命从信息技术逐渐转移到生命健康技术。 高通量测序等技术“读”DNA让遗传信息解读和应用快速普及,正在酝酿的“基因2.0时代”通过基因合成“写”DNA深度理解和设计生物系统,将变革育种、能源和环境等诸多领域。科学家们可以对编码基因、信号通路、代谢途径乃至基因组进行设计和合成。各国政府已将合成生物学技术作为重点资助对象,涉及合成生物学技术的创新创业也备受资本市场的青睐。
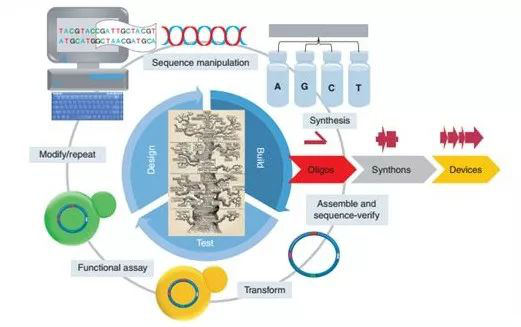
基因合成属于生物合成的范畴,具体包括计算机设计、DNA/RNA合成、CRISPR编辑等技术。在流程上是一个不断设计、改造、验证、修正的试错过程,以此来评估工程化改造生物系统的预期功能。 【基因合成流程】 1. 通过使用计算机软件设计DNA序列,这些DNA序列可构成生物电路或通路(图1)。 2. DNA序列被分割成更小的重叠片段(通常为200-1500bp的片段),然后进一步分割成可化学合成的一组单链寡核苷酸。 3. 单链寡核苷酸通过各种DNA组装技术,最终被组装成更大的DNA片段并进行克隆验证。 4. 验证后的DNA序列被转入细胞进行功能测试。 5. 根据功能试验结果,对本轮设计序列进行更改,并重复测试周期,形成迭代。
图1:基因合成的循环过程。(来源:Cold Spring Harb Perspect Biol)
二 基因合成的发展历程
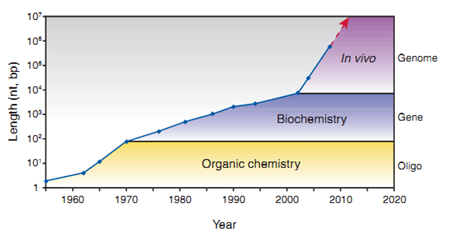
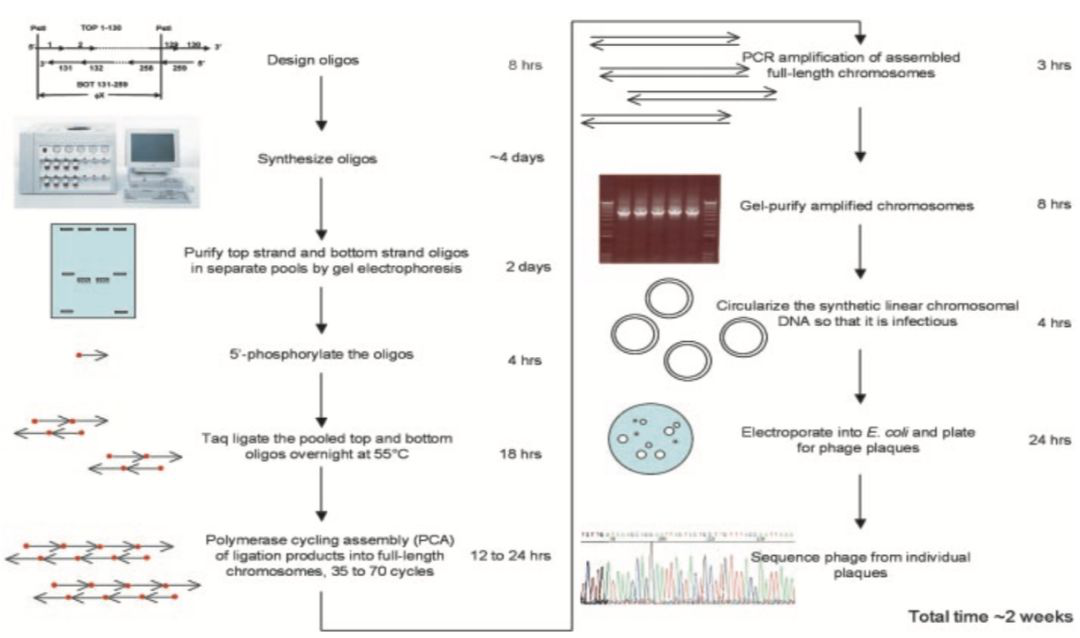
图2:DNA人工合成的发展。(来源:Nature Biotechnology) 基因合成在1970年前只是单链寡核苷酸链形式。自1970年后,双链DNA(>100 bp)的合成开始迅速发展。 2002年,脊髓灰质炎病毒(poliovirus)基因组(约7,500 kb)被成功合成(Cello, Paul et al. 2002),正式开启基因组合成时代。 2010年,Venter 和 Smith等将人工合成的蕈状支原体(Mycoplasma mycoides)基因组转入到山羊支原体(Mycoplasma capricolum)宿主细胞中(Gibson, Glass et al. 2010),走出了人工合成基因创造新细胞的历史性一步。 从 2011 年开始的酵母基因组合成计划(Sc2.0),当前已经完成了 2、3、5、6、10 和 12 号染色体的合成,其中中国科学家做出了重大贡献。DNA的合成与组装未来的发展也许会包括复杂的微生物群落或细胞组织的从头建立。 三 寡核苷酸的设计和合成流程 一条基因序列的合成起始于寡核苷酸的设计和化学合成,通过各种组装策略得到0.3-0.5 kb左右的短片段,再进行1-2 kb基因长度DNA序列的组装。 合成具有复杂结构的基因甚至基因组水平DNA时,合理的设计是关键。靶序列通常需要通过软件从头设计,包括密码子优化、DNA二级结构调整以及其他可能影响系统功能(如蛋白表达)的一系列理性设计。 【常用的DNA序列设计软件】 · DNAWORKS (http://mcl1.ncifcrf.gov/dnaworks/dnaworks2.html) · GEMS(http://software.kosan.com/GeMS) · Oligo Cuts · GeneDesign(http://slam.bs.jhmi.edu/gd/ · TmPrime(http://prime.ibn.a-star.edu.sg) 传统的寡核苷酸合成一般称为柱式合成,基于四步法亚磷酰胺化学合成方法(Froehler,Matteucci, 1983),在固相上进行寡核苷酸合成,现在仍被大多商业化DNA合成公司所采用。 该过程将酸活化的脱氧核苷亚磷酰胺分子耦合到一个固定在固相(一般是硅材质表面)的脱氧核苷酸分子上。在第一个合成循环中,核苷酸链从第一个固定在表面上受保护的核苷酸分子开始延伸。其中,经常采用的固定化表面主要是可控孔度玻璃( CPG)或者聚苯乙烯微珠(PS beads)。试剂被泵入并流经材质表面,诱导分步添加的核苷酸单体加入到寡核苷酸链上,使寡核苷酸链不断延长。 【每添加一个核苷酸单体到核苷酸链的四步过程】 (1)脱保护:一种弱酸从延长的核苷酸链的5’端末端移除二甲氧三苯甲基基团,并产生一个5’端活性羟基基团。 (2)耦合:脱氧核苷亚磷酰胺分子在适当的活化剂(如四唑,tetrazole)作用下,会产生一个活性单体分子,与上一步产生的5’羟基基团进行反应。 (3)盖帽:未耦合的5’羟基基团会被封闭,经常采用的封闭剂是酰化剂。 (4)氧化:连接核苷酸分子之间的亚磷酯键是不稳定的,容易被酸、碱水解,所以需要被氧化成为更加稳定的磷酸三酯。 整个合成过程完成后,采用浓氨水将寡核苷酸链从固相上切除,寡核苷酸链上的保护基团会被同时移除,残留最初固定在表面的那一个残基。
目前,基于亚磷酰胺合成法的DNA全自动合成仪已经可以将单次合成的通量从1根提高到1,536根序列(Cheng, Chen, Kao, et al., 2002)。亚磷酰胺方法合成寡核苷酸的成本也在逐渐降低。柱式合成通常以10至1000 nmol的规模合成寡核苷酸,每个碱基的成本在0.05至0.17美元之间。这些寡核苷酸通常可以合成多达100nt,错误率在0.5%或以下,每个单体耦合效率通常可达99%。 然而,随着寡核苷酸长度的增加,全长寡核苷酸的产量通常会下降。并且,合成寡聚物的随机去嘌呤会影响全长寡核苷酸的产量,特别是在合成周期的酸脱保护步骤中,腺嘌呤容易受到去嘌呤影响,并最终促使寡核苷酸主干的断裂,从而降低全长寡核苷酸的产量。对于较长的寡核苷酸合成来说,该问题尤其突出(Efcavitchand Heiner 1985;Septak 1996;LeProust et al.2010)。 寡核苷酸合成质量的进一步降低是由于在合成过程中引入随机突变,主要是单碱基缺失类型。这类错误主要来自DMT保护基团的不完全去除或合成周期中耦合和封盖步骤的综合效率低下。完全去除合成错误是不太可能的,因为任何化学反应的效率都无法达到100%。但合成工艺的改进仍然能提高寡核苷酸的合成长度和质量(LeProust et al.2010年)。 当前主流的DNA合成供应商一般是基于柱式合成的寡核苷酸。因为柱式寡核苷酸成分单一,产量高,错误率低,对于基因合成来说组装更容易,保真性也更高,这就极大降低了操作的难度和纠错的成本。但柱式合成存在显而易见的缺陷,比如合成通量低、成本高,无法支持大规模基因的合成。
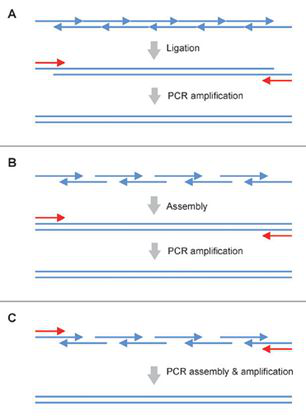
四 基因合成的技术流程 人工合成DNA一般起始于预先合成的寡核苷酸(一般60-120 nt)。虽然更长的核苷酸链(长至300-600 nt)可以直接合成,但是DNA的化学合成随着长度延长,合成产率会急剧降低,因此直接合成长链DNA的方法并没能得到推广。 在过去几十年里,将寡核苷酸组装成为基因长度DNA的方法在不断发展。其中基于连接酶的组装和基于聚合酶链式反应的组装是最主要的两种方法(图3)。
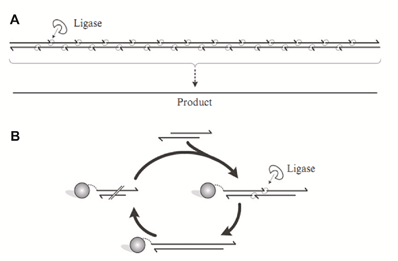
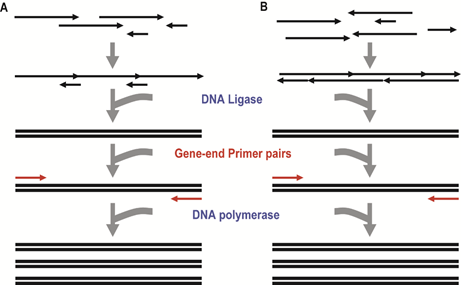
图3:酶介导的基因合成。(来源:Molecular Biosystems) 1) 基于DNA连接酶的组装方法 最早的依赖连接反应的基因合成可以追溯到1960s后期,Gupta等人使用寡核苷酸合成技术和DNA连接酶介导的组装技术,合成了一条30bp的酵母丙氨酸tRNA基因片段。二十年后,第一条编码人胰岛素A的基因(63bp)被人工合成出来,合成方法就基于DNA连接酶。八十年代一种称为“鸟枪法连接(Shotgun Ligation)”的新型组装方法开发出来。这种方法将目标基因切割成为多条片段,相邻片段之间存在重叠区域,而每条片段由多条磷酸化的寡核苷酸组成(如图4)。
图4:连接酶介导的基因合成方法。(来源:Synthetic Biology)
随着耐热性DNA连接酶(Thermostable DNA ligase)的发现,基于LCR的基因组装方法变得更加方便易行。使用耐热连接酶的一个显著优势在于连接时的较高温度(约50-60℃)可以减弱二级结构的形成,从而提升组装成功率;并且,寡核苷酸在高温条件下的退火及连接可以提高正确配对的比例,提高连接的准确度。1990年Smith课题组使用搭桥式连接(图5A),构建了一条924 bp的编码辣根过氧化物酶同工酶的基因。2003年Smith课题组应用LCR-PCR思路,成功地在两周内完成噬菌体Phi X174 (5386 bp)基因组的人工合成并验证其功能(如图6)。
图5:基于LCR-PCA的DNA组装方法。(来源:作者)
图6:phi X174噬菌体基因组(5386 bp)的从头合成流程示意图。(来源:Proc Natl Acad Sci USA)
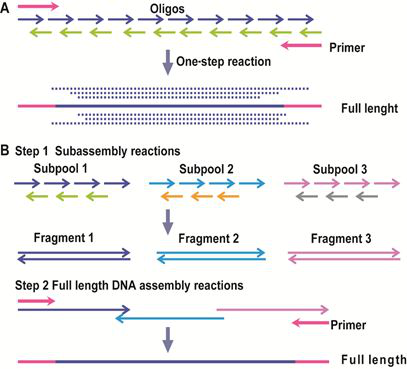
2) 基于DNA聚合酶的组装方法 聚合酶链式反应(PCR)是一种最常用的基于寡聚核苷酸的基因组装方案。通过一步PCR组装的方法,可以将一组寡核苷酸混合物一步组装成为基因长度DNA,但这种方法得到的PCR产物通常是一组长度不一的DNA(如图7A)。 另外一种基于PCR的两步法组装方法,首先将基因拆分成一系列亚库(subpools/ subassembly reactions),分别组装成为片段,再将这些片段混合,最终组装成为完整基因(如图7B)。
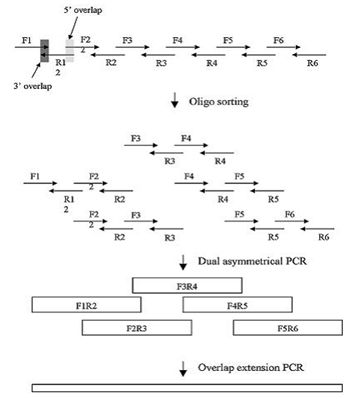
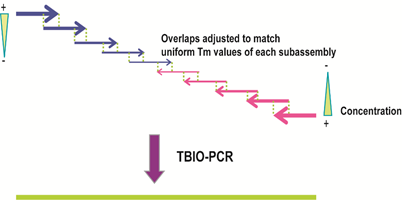
图7:基于PCR的DNA组装方法。(来源:作者) Stemmer等人在1995年报道了基于PCR原理的、由寡核苷酸一步法组装全长基因的方法(Stemmer, Crameri et al. 1995)。随着目标基因结构复杂性的提升和长度增长,寡核苷酸的合成数量在不断攀升。为了提高组装的成功率,提高通量并降低出错率,DNA的组装技术也需要不断改进。Young和Dong等人结合了不对称PCR(Dual asymmetrical PCR, DA-PCR)(Sandhu, Aleff et al. 1992)和重叠PCR(Overlap extension PCR, OE-PCR)(Horton, Hunt et al. 1989)技术,开发了一种新的PCR组装方法(图8)。 在设计时,片段之间的重叠区域长度大于寡核苷酸之间的重叠区域。当外侧的引物浓度高于寡核苷酸浓度时,长的亚组装PCR产物(DA-PCR产物)会被优先选择扩增出来。每组DA-PCR产物可以通过OE-PCR方法延伸得到全长产物。使用多个亚组装(前组装)反应可以降低寡核苷酸混合物之间的非特异性退火,从而提高组装产物的纯度。 Gao等人在2003年发明了热动力学平衡法 (thermodynamically balanced inside-out, TBIO),开创了一种新的依赖PCR的DNA拼装技术(Gao, Yo et al. 2003),主要原理是调整寡核苷酸组装过程中单链寡核苷酸之间及它们和引物之间的热力学平衡。该方法的设计及合成过程包括五个主要步骤,尤其重要的是引物设计和组装策略。在该策略中,正义链的寡核苷酸覆盖了基因N-端的一半序列,而反义链上的寡核苷酸则覆盖了C-端的一半序列。两组寡核苷酸会在基因或者片段的中心相遇(图9)。利用TBIO方法,4-6对约60 nt长的寡核苷酸可以用来拼装一段约400-500 bp长的基因片段。TBIO拼装过程可以重复进行,直到得到所需的全长DNA。Gao等人使用TBIO-PCR拼装方法得到的15条基因序列的错误率是0%-0.3% (Gao, Yo et al. 2003)。
图8:基于DA-PCR和OE-PCR的DNA组装方法。(来源:Nucleic Acids Research)
图9:TBIO-PCR方法拼装DNA的原理示意图。(来源:作者) Xiong等人在2004年描述了另外一种基于PCR的组装技术——两步法DNA合成方法(PCR-based, two-step DNA synthesis, PTDS),成功合成了1,230 bp长的Peniophora lycii phytase基因(Xiong, Yao et al. 2006)和5,367 bp长的CrtEBWY基因(Xiong, Yao et al. 2004)。而该方法的改进版本,引进了对寡核苷酸的胶回收步骤和基于OE-PCR的错误纠正步骤,可以提高合成产物的整体保真性(Xiong, Yao et al. 2006)。 3) 组装方法的选择 连接介导的DNA拼装方法和PCR介导的拼装方法各有利弊,对大多数DNA序列来说,两种方法都可以实现从寡核苷酸到全长基因的拼装。在PCR介导的组装方法中,双链中任意一条链上的相邻寡核苷酸(属于两条链或者同一条链)之间都可能存在核苷酸的缺失,这在连接介导的组装过程中则不会出现。这一特点使得PCR介导的组装方法在合成大量DNA时稍具成本优势。 PCR介导的一步组装方法的优势是快速、方便。但是,并不是所有序列都可以通过PCR的方法组装得到。在合成一些具有特殊结构,如重复序列或复杂二级结构的DNA序列时,热稳定性连接酶介导的基因拼装方法是更好的选择。另外,相比连接反应,全部由PCR过程构成的拼装方法可能会引入较多额外的错误。化学合成的寡核苷酸的错误率一般是1-3 errors/kb (Hoover and Lubkowski 2002, Binkowski, Richmond et al. 2005),基于该保真水平的寡核苷酸为材料,通过PCR方法拼装得到的DNA序列的错误率会提高一个数量级(Binkowski, Richmond et al. 2005)。因此,所有PCR拼装策略都需要高质量的寡核苷酸。在许多例子中,拼装长链DNA使用的寡核苷酸都是商业提供并且经过胶纯化后的产物(Wu, de Kievit et al. 2004, Bi, Zhou et al. 2005)。
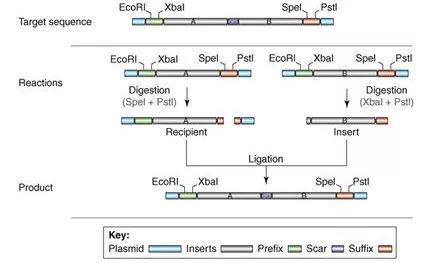
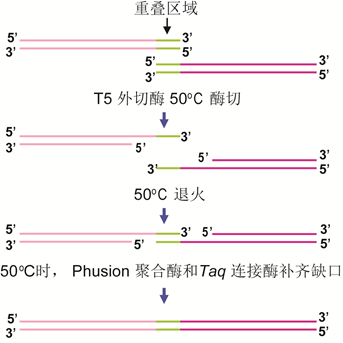
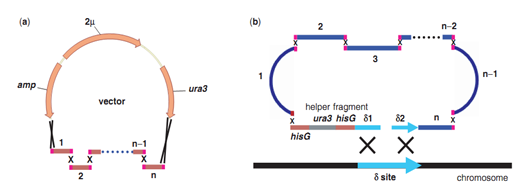
五 基因组合成和基因组编辑技术 随着长片段DNA组装技术的不断进步,出现了构建大型酶复合物(Kodumal et al. 2004)、整个代谢通路(Temme et al. 2012)甚至整个基因组的方法(Smith et al. 2003;Gibson et al. 2008a)。体外组装和体内组装法共同推进更大的DNA结构的人工合成。 体外组装包括传统酶切连接、BioBrickTM组装法(图10)、Gibson组装(图11)等。其中,Gibson组装可能是将多个DNA片段组装成更大的结构最常用的的方法(Gibson et al. 2010)。该方法采用一锅等温技术,利用含有热稳定DNA聚合酶、DNA连接酶和外切酶的酶混合物,变性、退火和修复相邻重叠的DNA序列,组装所需的结构。该方法被用来直接以60 mer长度寡核苷酸组装了16.3 kb的小鼠线粒体基因组(Gibson et al. 2010)。 利用酿酒酵母的同源重组能力,还可以在体内高效组装更大的合成DNA片段(图12)。例如,酵母可以将外源DNA从相互重叠的片段组装成更大的DNA结构,J. Craig Venter Institute的研究人员已经成功地使用酵母组装了多个0.5-1 Mb的细菌基因组(Gibson et al. 2008a),甚至直接以相互重叠的寡核苷酸进行组装 (Gibson 2009, 2011b, 2012)。
图10:BioBrickTM拼装长链DNA的原理示意图。(来源:Trends in Biotechnology)
图11:外切酶介导的长链DNA拼装方法(Gibson法)的原理示意图。(来源:作者)
图12:基于酵母体内重组原理的一步法拼接代谢通路。(来源:Nucleic Acids Research)
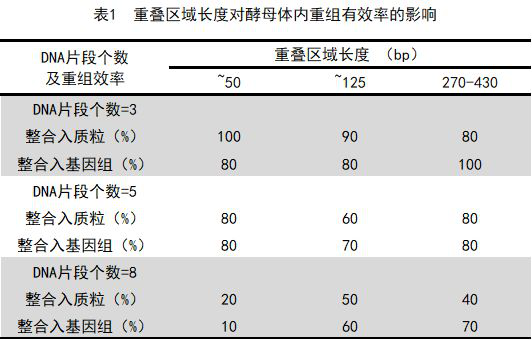
表1:重叠区域长度对酵母体内重组有效率的影响(来源:Nucleic Acids) 基因组完成组装后,需要依托基因组编辑技术进行验证并进行部分校正。
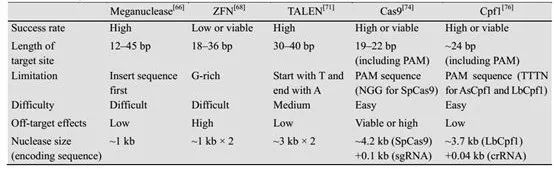
表2:不同核酸酶的基因编辑体系的比较(来源:生物工程学报) 基因组编辑技术包括Cre-loxP重组酶系统、位点特异性切割蛋白(如锌指蛋白核酸酶ZFN、类转录激活因子效应物核酸酶TALEN,以及CRISPR-Cas9技术)。简单来说,工作模式是先切割后修复,并引入外源序列,达到基因组编辑的功能。
作者介绍
李璐璐 博士 李璐璐(微信号:luannelee),博士,毕业于中国科学技术大学生物化学与分子生物学专业,师从合成生物学先驱高晓连教授,主要进行基于芯片寡核苷酸的高保真组装技术开发,成果发表于Nucleic Acids Research 杂志。2014年加入杭州联川生物,现任研发部主管,负责DNA合成技术及NGS基因检测技术的开发工作。
参考资料: 1. Bi, L. J., Y. F. Zhou et. al., (2005). "Construction and characterization of different MutS fusion proteins as recognition elements of DNA chip for detection of DNA mutations." Biosens Bioelectron 21(1): 135-144. 2. Binkowski, B. F., K. E. Richmond et. al., (2005). "Correcting errors in synthetic DNA through consensus shuffling." Nucleic Acids Research 33(6) 3. Gao, X. X., P. Yo et. al., (2003)."Thermodynamically balanced inside-out (TBIO) PCR-based gene synthesis: a novel method of primer design for high-fidelity assembly of longer gene sequences." Nucleic Acids Research 31(22) 4. Gibson, D. G., J. I. Glass et. al., (2010). "Creation of a Bacterial Cell Controlled by a Chemically Synthesized Genome." Science 329(5987): 52-56. 5. Hoover, D. M. and J. Lubkowski (2002). "DNAWorks: an automated method for designing oligonucleotides for PCR-based gene synthesis." Nucleic Acids Research 30(10). 6. Horton, R. M., H. D. Hunt et. al., (1989)."Engineering Hybrid Genes without the Use of Restriction Enzymes - Gene-Splicing by Overlap Extension." Gene 77(1): 61-68. 7. Sandhu, G. S., R. A. Aleff and B. C. Kline (1992). "Dual Asymmetric Pcr - One-Step Construction of Synthetic Genes." Biotechniques 12(1): 14-16. 8. Stemmer, W. P. C., A. Crameri et. al., (1995). "Single-Step Assembly of a Gene and Entire Plasmid from Large Numbers of Oligodeoxyribonucleotides." Gene 164(1): 49-53. 9. Wu, Y., P. de Kievit, L. Vahlkamp et. al., (2004). "Quantitative assessment of a novel flow-through porous microarray for the rapid analysis of gene expression profiles." Nucleic Acids Research 32(15). 10. Xiong, A. S., Q. H. Yao, R. H. Peng et. al., (2004). "A simple, rapid, high-fidelity and cost-effective PCR-based two-step DNA synthesis method for long gene sequences." Nucleic Acids Research 32(12) 11. Xiong, A. S., Q. H. Yao, R. H. Penget. al., (2006). "High level expression of a synthetic gene encoding Peniophora lycii phytase in methylotrophic yeast Pichia pastoris." Applied Microbiology and Biotechnology 72(5): 1039-1047. 12. Hughes, R.A. and A.D. Ellington, Synthetic DNA Synthesis and Assembly: Putting the Synthetic in Synthetic Biology. Cold Spring Harb Perspect Biol, 2017. 9(1) 13. Singh-Gasson, S., et al., Maskless fabrication of light-directed oligonucleotide microarrays using a digital micromirror array. Nature Biotechnology, 1999. 17: p. 974. 14. Gao, X., et al., A flexible light-directed DNA chip synthesis gated by deprotection using solution photogenerated acids. Nucleic Acids Res, 2001. 29(22): p. 4744-50. 15. Tian, J., et al., Accurate multiplex gene synthesis from programmable DNA microchips. Nature, 2004. 432(7020): p. 1050-4. 16. Zhou, X., Microfluidic PicoArray synthesis of oligodeoxynucleotides and simultaneous assembling of multiple DNA sequences. Nucleic Acids Res., 2004. 32: p. 5409-5417. 17. Quan, J., et al., Parallel on-chip gene synthesis and application to optimization of protein expression. Nat Biotechnol, 2011. 29(5): p. 449-52. 18. Kosuri, S. and G.M. Church, Large-scale de novo DNA synthesis: technologies and applications. Nature Methods, 2014. 11: p. 499. 19. Richmond, K.E., Amplification and assembly of chip-eluted DNA (AACED): a method for high-throughput gene synthesis. Nucleic Acids Res., 2004. 32: p. 5011-5018. 20. Lubock, N.B., et al., A systematic comparison of error correction enzymes by next-generation sequencing. Nucleic acids research, 2017. 45(15): p. 9206-9217. 21. Fodor, S.P., et al., Light-directed, spatially addressable parallel chemical synthesis. Science, 1991. 251(4995): p. 767-73. 22. Pease, A.C., et al., Light-generated oligonucleotide arrays for rapid DNA sequence analysis. Proc Natl Acad Sci U S A, 1994. 91(11): p. 5022-6. 23. Egeland, R.D., F. Marken, and E.M. Southern, An electrochemical redox couple activitated by microelectrodes for confined chemical patterning of surfaces. Anal Chem, 2002. 74(7): p. 1590-6. 24. Ghindilis, A.L., et al., CombiMatrix oligonucleotide arrays: genotyping and gene expression assays employing electrochemical detection. Biosens Bioelectron, 2007. 22(9-10): p. 1853-60. 25. Recht, M.I., et al., RNA sequence determinants for aminoglycoside binding to an A-site rRNA model oligonucleotide. J Mol Biol, 1996. 262(4): p. 421-36. 26. Hughes, T.R., et al., Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer. Nat Biotechnol, 2001. 19(4): p. 342-7. 27. 李诗渊、赵国屏、王金.(2017)合成生物学技术的研究进展——DNA合成、组装与基因组编辑,生物工程学报 33 (3):343-360 |

更有优质直播、研选好物、福利活动等你来!