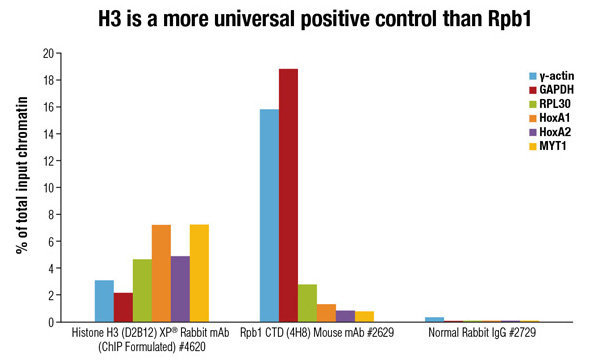
我用冷泉港提供的protocol(http://cshprotocols.cshlp.org/cgi/content/full/2009/9/pdb.prot5279#R42)
1. Add 27 μL of 37% formaldehyde per milliliter of cell culture (in growth medium on tissue culture plates or in tissue culture flasks) while slowly shaking the cells for 10 min at room temperature.
The final concentration of formaldehyde is 1%.
2. Quench cross-linking by adding 100 μL of 1.375 M glycine per milliliter of culture.
3. Treat the cells as follows:
For adherent cells
i. Remove the growth medium and dispose of it appropriately.
ii. Wash cells twice with 10 mL of cold 1X PBS, and then scrape the cells into 1X PBS in a 15-mL conical tube.
iii. Pellet the cells at 1500 rpm for 10 min at 4°C.
For nonadherent cells
iv. Transfer the cells to a 15-mL conical tube.
v. Pellet the cells at 1500 rpm for 10 min at 4°C.
vi. Wash the cells twice with 10 mL of cold 1X PBS.
4. Resuspend the cell pellet in 10 mL of cell lysis buffer for ChIP (cold) and incubate for 10 min on ice.
5. Pellet the cell nuclei at 1000 rpm in a benchtop centrifuge for 10 min at 4°C. Aspirate the supernatant carefully.
7. Resuspend the nuclear pellet in 1 mL of nuclei lysis buffer for ChIP supplemented with protease inhibitors and incubate for 10 min on ice.
8. Proceed with sonication. Ensure that the samples do not foam and are kept as cold as possible.
在完成1-8步骤后用
1. mix 20 ul extract + 1ul 10% SDS + 0.5 ul proteinaseK
2. incubate 1hr at 37 degrees C followed by 2 hrs at 65 degrees C
3. phenol/chloroform extract the samples
4. chloroform extract the samples
5. EtOH ppt DNA with 10 ug/ml glycogen (final concentration)
6. resuspend DNA in 20 ul TE
7. treat with 40 ug/ml RNAse for 1hr at 37 degrees C
8. run half of each sample on a 1.5% agarose gel with EtBr with an appropriate DNA sizing ladder (sheared DNA should run between 100 bp and 1000 bp)
步骤提取DNA后电泳没有DNA条带是怎么回事呢?