| 实验所需「材料」、「试剂」、「耗材」具体见「其他」 1. 乙醇沉淀 50 ug 总 RNA,重溶于 15.5 ul 无 RNase 的水中。加入 1.5 ul 10 umol/L cDNA 引物 CP29V,65°C 变性 5 min(如在一个加热块上),冰上冷却。
2. 在冰上混合第一链合成反应的成分(总体积 29.1 ul): 17.0 ul 刚刚变性的 cDNA 和 cDNA 引物 3.0 ul 100 mmol/L 无 RNase 的 DTT 6.0 ul 5× SuperScript 缓冲液 1.5 ul 10 mmol/L 无 RNase 的 dNTP 0.6 ul 40 U/ul 的 RNase 抑制剂(如 RNasin) 1.0 ul 200 U/ul SuperScript II 逆转录酶 混匀,42°C 温育 1h。置于冰上中止反应。
3.在冰上混合第二链合成反应的成分(总体积 207.2 ul): 48 ul 5× 第二链缓冲液 II 3.6 ul 10 mmol/L dNTP 148.4 ul H2O 1.2 ul 1.5 U/ul RNase H 6.0 ul 10 U/ul E.coli DNA 聚合酶 I
4. 把第一链的和第二链反应体系合在一起。混匀,22°C 温育 2 h。完成第二链合成后, 75°C 加热 20 min 灭活 DNA 聚合酶 I。
5. 用 100 ul pH 8.0 的 TE 缓冲液平衡的酚抽提。再用 100 ul 氯仿抽提。
6.为进行按分子大小选择性的 PEG 沉淀,小心混合: 200 ul 酚/氯仿抽提的 ds cDNA 1.0 ul 20 mg/ml 糖原 200 ul 28%(m/V)PEG 8000/3.6 mmol/L MgCl2 反应体系(总体积 401 ul)室温放置 5 min,然后 10°C,最高速离心 15 min。70% 的乙醇小心洗涤沉淀。
这步沉淀可除去未掺入的 cDNA 引物和小分子核酸(如低于 100 nt)。因为 PEG 大小选择性沉淀易受微小浓度变化的影响,所以必须遵循如下方针:
6.1 确保准确吸取 200 ul ds cDNA。溶解在水相中的氯仿的蒸气压力倾向于取代吸头中液体的体积使得准确的吸取变的困难。克服这个问题的一个办法是在吸取 200 ul 体积前,先用 50~100 ul 的体积反复吹吸(5~10 次)氯仿饱和的水相,使吸头中的氯仿蒸气达到饱和。
6.2 因为 28% 的 PEG 8000/3.6 mmol/L MgCl2 相当黏稠,要小心地慢慢吸取,同样需要确保准确转移所需的体积。
6.3 首先小心地重复颠倒混勻,然后剧烈地涡旋混匀。因为溶液黏稠,彻底混匀需要一些时间。
7. 在冰上把沉淀溶解在下述溶液里(总共 96 ul): 15.0 ul 10× 通用缓冲液 81.0 ul H2O
8. 加入 4 ul 4 U/ul 的 MboI,37°C 温育 1 h。65°C 加热 20 min 使酶灭活。
9. 用 50 ul pH 8.0 TE 平衡的酚抽提,再用 50 ul 氯仿抽提。加入 1 ul 糖原和 10 ul pH 5.2 的 3 mol/L 乙酸钠,2.5 倍体积 100% 的乙醇。最高速离心 20 min, 70% 乙醇洗涤沉淀。轻微晾干(5~10 min)。不要加热或真空干燥,因为过度的干燥会使得 DNA 沉淀很难溶解。
10. 把沉淀溶解在由以下成分组成的连接混合液里(总体积 20 ul) 1.2 ul 10× 连接缓冲液 2.0 ul 10 mmol/L ATP 8.0 ul 0.5 ug/ul MboI-连接子 ML2025 7.8 ul H2O 1.0 ul 1 U/ul 的 T4 DNA 连接酶 16°C 连接过夜或 4°C 连接一个周末。
11. 加入 90 ul 的水,混匀,用 50 ul TE 平衡的酚抽提,再用 50 ul 氯仿抽提。配制第二次 PEG 沉淀反应去除未连接的连接子(总体积 201 ul): 100 ul 酚抽提过的连接产物 1.0 ul 糖原 100 ul 28% 的 PEG 8000/3.6 mmol/L MgCl2 室温放置 5 min,然后 10°C,最高速离心 15 min。70% 的乙醇小心洗涤沉淀,重溶于 40 ul pH 8.0 TE 缓冲液。
12. 配制第一轮扩增反应。在冰上把 3 个 4 umol/L CP28X1(X1=A、C 或 G)引物和 4 个 4 umol/L 引物 ML19Y1(Y1=A、C、G 或 T)各 1 ul 两两混合在分开的管里(共 12 个反应)。用所有其他的成分配制一个通用混合液(一个反应的配方): 2.0 ul 模板(PEG 沉淀的连接产物) 2.0 ul 10× PCR 缓冲液 1.5 ul 20 mmol/L MgCl2 0.4 ul 10 mmol/L dNTP 2.0 ul RediLoad 9.9 ul H2O 0.2 ul 5 U/ul Taq DNA 聚合酶 配制好反应后把管子放到热循环仪上预热到 90°C。
13. 运行下面的循环程序: 起始步骤: 1 min 94°C(变性) 25 轮循环: 20 s 94°C(变性) 30 s 65°C(引物复性) 4 min 72°C(引物延伸) 最后步骤: 不确定 10°C(保持/延伸)
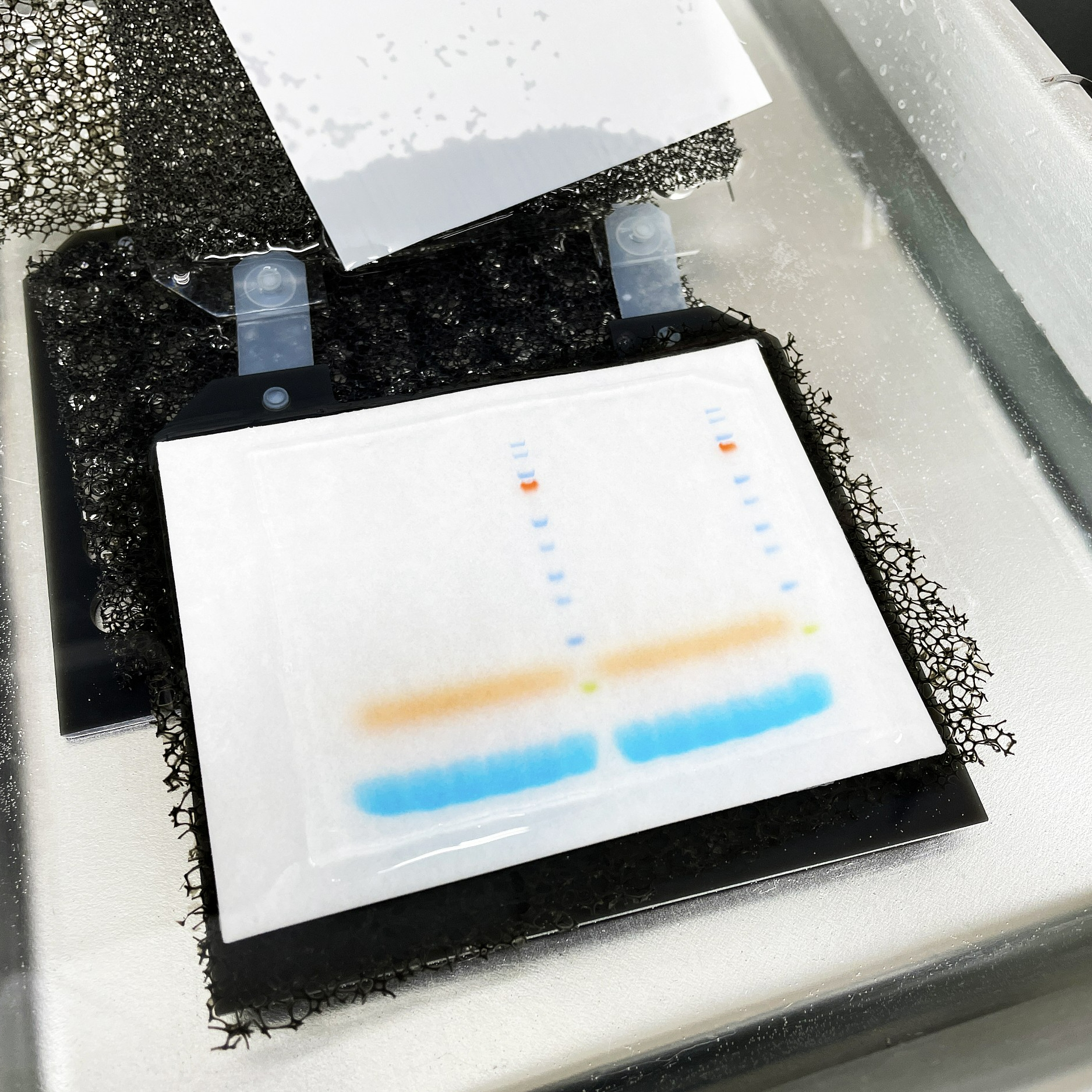
14. 每个反应取 10 ul 上样到 1.5% 的琼脂糖凝胶上,电泳检查是否扩增成功。100 bp 的序列阶梯作为分子质量对照。 引物量决定产物的量,可以以此调整 PCR 的条件。长时间的延伸确保不同大小的产物都能得到扩增而没有不利于长片段的偏差。琼脂糖电泳应该出现约 100~700 bp 的拖尾,少有明确的条带(如果有的话)。最重要的是用相同引物扩增的不同 RNA 样品的扩增产物应该看起来基本一样。 如果出现不同或量不一样,说明扩增前某一步的酶操作效率可能是太低了。
15. 用 pH 8.0 0.25× 的 TE 缓冲液按 1:100 稀释反应产物。
16. 配制第二轮扩增反应。在两块 96 孔板上两两混合 12 个 CP28X1 X2 引物和 16 个 MLDY1Y2 引物(每个样品 192 个不同的反应;每个 20 ul): 2.0 ul 模板(稀释的第一轮扩增产物) 2.0 ul 10× PCR 缓冲液 1.5 ul 20 mmol/L MgCl2 0.4 ul 10 mmol/L 标准 dNTP 2.0 ul 4 umol/L 引物 CP28 X1X2(X2=A、C、G 或 T) 2.0 ul 4 pmol/L 标记的引物’MLlSY1Y2(Y2=A、 C、G 或 T) 2.0 ul RediLoad 7.9 ul H2O 0.2 ul 5 U/ul Tag DNA 聚合酶 确保每个反应,第一轮和第二轮使用的一致的 X1 和 Y1。
17. 运行步骤 13 的程序,但只要 20 个循环。用琼脂糖凝胶电泳检查是否扩增成功 (也见步骤 13)。
18. 每个反应转移 5 ul 到新装有 5 ul 甲酰胺缓冲液的 96 孔板上。75°C 变性 2 min。
19a. 放射性标记样品:取 1~2 ul 上样于 6% 的变性聚丙烯酰胺凝胶电泳。
19b. 非放射性标记使用直接印迹电泳。 |