大家都在搜


| 实验材料 | |
|---|---|
| 试剂、试剂盒 | |
| 仪器、耗材 | |
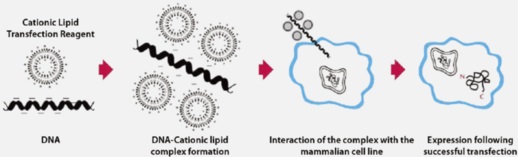
| 实验步骤 | 材料 缓冲液与溶液 贮存液、缓冲液与试剂的成分见附录 1。 稀释贮存液至所需浓度。 CaCl2(2.5mol/L) 氯喹(100 mmol/L)(选用) 溶解 52 mg 二磷酸氯喹于 1 ml 去离子水,用 0.22um 的滤器过滤除菌。用箔纸包裹好存放滤液的试管,-20°C 保存。见步骤 5。 Giemsa 染液(10%m/V) Giemsa 染液应是使用前用磷酸盐缓冲液或水新制备的,用 Whartman1 号滤纸过滤。 甘油(15%V/V)1XHEPES 盐缓冲液溶解(备选) HEPES 盐溶液过滤除菌,使用前加入高压灭绝的 15% 的甘油。见步骤 5。 2XHEPES 盐缓冲液 140 mmol/LNaCl 1.5 mmol/LNa2 HPO4•2 H20 50 mmol/L.HEPES 于 90 ml 蒸馏水中溶解 0.8 gNaCl,0.027 gNa2 HPO4•2 H20,1.2 gHEPES, 用 0.5mol/LNaOH 调节 pH 至 7.05, 用蒸馏水将体积补至 100 ml, 用 0.22um 的滤器过滤除菌;分装成 5 ml 等份,-20°C 保存,1 年内使用。 甲醇 磷酸盐缓冲液 溶液使用前过滤除菌. 室温保存。 丁酸钠(500 mmol/L)(备选) 在化学通风橱内,用10mol/LNaOH 调节丁酸贮存液至 pH7.0。用 0.22um 的滤器过滤除菌;分装成1 ml 等份,-20°C 保存,见步骤 5。 0.1XTE(PH7.6) 1 mmol/LTrLi-Cl(pH7.6) 0.1 mmol/LEDTA(pH7.6) 用0.22um的滤器过滤除菌,分装,4°C 保存。 核酸与寡梭苷酸 质粒 DNA 用 0.1XTE(pH7.6) 溶解 DNA 至终浓度 25ug/ml; 每毫升培养基需要 50ul 质粒溶液。质粒 DNA 应用层折柱(见第 1 章方案 9) 或 CsCl-溴化乙锭梯度离心(见第 1 章方案 10) 纯化以获得最高转化效率。如果质粒 DNA 起始有限,可加入载体 DNA 将终浓度调至 25ug/ml。实验室内制备的真核运载 DNA 的转染效率通常比购买的 DNA 如小牛胸腺或鲑精 DNA 的转染效率高。运载 DNA 使用前通过乙醇沉淀或氯仿抽提除菌。 培养基 细胞生长培养基 [完全培养基与(优化备选)选择性培养基] 特殊设备 组织培养皿(60 mm) 或 12 孔板 本方法适用于 60 mm 组织培养皿或 12 孔板培养细胞。如果使用其他多孔板,细胞瓶或其他直径的培养皿,按比例改变细抱浓度与试剂的用量。见表 16-3。 辅助试剂 步骤 6 所需试剂见第 6 章方案 10, 第 7 章方案 8。 细胞与组织 指数生长的哺乳动物细胞 方法 1. 转染前 24 h,通过胰酶消化收集细胞,用适当的完全培养基以 1x105 至 4x105 细胞/cm2 的密度平铺细胞于 60 mm 组织培养皿或 12 孔板上。于含 5%~7%CO2 的 37°C 温箱孵 20~24 h。转染前1h 换液。 为得到较高的转染效率,注意使用指数生长的细胞,转染时细胞的汇合度不能超过 80%。 2. 按照下述方法制备磷酸钙-DNA 沉淀:于 5 ml 灭菌塑料管内混合 100ul 2.5mol/LCaCl2 与 25ug 质粒 DNA,如有必要用0.1xTE(pH7.6) 将体积补至 1 ml。室溫下将以上 2x 钙-DNA 溶液与等体积的 2xHEPES 盐溶液混合。迅速弹敲试管侧壁混匀溶液,静置 1 min。 如果转染大量细胞,可以将沉淀反应混合物的体枳扩大至两倍或 4 倍,通常,培养皿、孔或细胞瓶内毎 1 ml 培养基加 0.1 ml 磷酸钙-DNA 沉淀物。 3. 立即将磷酸钙-DNA 悬液转移至上述单层细胞的细胞培养基中。每 1 ml 培养基加 0.1 ml 悬液。轻轻摇动平皿混勻培养基,培养基会变成混浊的橘黄色。一旦形成 DNA 沉淀,转染效率会大大降低,因此此步动作应尽可能迅速。如果是用氯喹、甘油与/或丁酸钠处理细胞,直接进行步骤 5。 有时候,先去掉培养基再直接加入磷酸钙-DNA 于暴露的细胞上会得到较髙的转染效率。然后,室温下孵育细胞 15 min, 再加入培养基。 4. 如果转染的细胞不用转染促进剂处理,则置于含 5%~7%CO2 的 37°C 温箱孵育。2~6 h 后,吸去培养基与 DNA 沉淀。加人 5 ml 37°C 预热的完全培养基,将细胞放回孵箱孵育 1~6 天。继续步骤 6 检测转染 DNA 的瞬时表达,或者如果是稳定转化则直接进行步骤 7。 5. 在磷酸钙-DNA 沉淀物的存在下用氯喹处理细胞或者吸去共沉淀溶液后将细胞暴露于甘油与丁酸钠中会促进细胞吸收 DNA。 用氯喹处理细胞 氯喹是一种弱碱,推测是通过抑制细胞内溶酶体水解酶降解 DNA 而发挥作用 (Luthman and Magnusson 1983)。细胞对氯喹毒性的敏感度限制了培养基中加入氯喹的浓度及时间,不同细胞所需氯喹最佳浓度根据经验而定(见信息栏二磷酸氯喹)。 a. 在把磷酸钙-DNA 沉淀物加入细胞前或后按 1:1000 将 100 mmol/L 氯喹直接加入培养基中。 b. 细胞于含 5%~7%CO2 的 37°C 温箱中孵育 3~5 h。 大多数细胞可在氯喹中生存 3~5 h。氯喹处理的细胞多呈泡状。 c. 在用 DNA 与氯喹孵育细胞后,移去培养基,用磷酸盐缓冲液洗涤细胞,加 5 ml 预热的完全培养基。细胞于孵箱中培养 1~6 天。继续步骤 6 检测转染 DNA 的瞬时表达,或者直接进行步骤 7 以获得稳定转化子。 用甘油处理细胞 此操作可在氯喹处理后进行。由于细胞对甘油毒性的敏感度不同,应在使用前先测试最佳处理时间(30s 至 3 min)。 a. 在把磷酸钙-DNA 沉淀物加入细胞 2~6 h 后(±氯喹),吸去培养基,磷酸盐缓冲液洗涤细胞层。 b. 每平皿细胞加 1.5 ml 15% 甘油 1xHEPES 盐缓冲液,依据事先测试的最佳时间于 37°C 孵育细胞。 c. 吸去甘油,磷酸盐缓冲液洗涤细胞一次。 d. 加 5 ml 预热的完全培养基。细胞于孵箱中培养 1~6 天。继续步骤 6 检测转染 DNA 的瞬时表达,或者直接进行步骤 7 以获得稳定转化子。 丁酸钠处理细胞 丁酸钠的作用机制并不确定;但它是组蛋白脱乙酰基抑制剂(Lea and Randolph 1998), 推测丁酸钠会导致组蛋白过乙酰化形成使外来质粒 DNA 趋于转录的染色质结构(Workman and Kingston 1998)。 a. 甘油休克处理后,将 500 mmol/L 丁酸钠直接加入生长培养基(甘油处理的步骤 d)。细胞类型不同,所用丁酸钠的浓度也不同。例如: CV-1 10 mmol/L NIH-3T 37 mmol/L HeLa 5 mmol/L CHO 2 mmol/L 其他细胞系所需浓度依经验而定。 b. 细胞于孵箱中培养 1~6 天。继续步骤 6 检测转染 DNA 的瞬时表达,或者直接进行步骤 7 以获得稳定转化子。 6. 若要检测细胞转染后导入 DNA 的瞬时表达,可在转染后 1~6 天收获细胞。杂交分析 DNA 或 RNA。通过体内代谢标记进行放射免疫、免疫印渍、免疫沉淀或测定细胞提取物的酶活性来分析新合成蛋白。 为减小不同培养皿之间转染效率的差异,最好(1)用每一构建体转染数个培养皿;(2)孵育 24 h 后用胰酶消化细胞;(3)将细胞汇集起来;以及(4)重铺细胞于数个培养皿上。 7. 分离稳定转染体 a. 用非选择性培养基中孵育细胞 24~48 h, 使转染的 DNA 有足够时间表达。 b. 胰酶消化细胞并重铺细胞于选择性培养基中,或者直接加选择性培养基。 c. 每 2~4 天更换培养基,持续培养 2~3 周,目的是清除死细胞残骸,促进抗性细胞生长。 d. 克隆独立菌落、繁殖,以用于检测(方法见 Jakoby and Pastan 1979 或 Spector et al.1998b[《细胞实验手册》的第 86 章])。 e. 用预冷的甲醇固定细胞 15 min, 然后室溫下用 10%Giemsa 染色 15 min, 流水冲洗,这样可以纪录细胞克隆数目。 根据稳定转染效率确定转染细胞的稀释度,重铺细胞产生独立克隆,不同细胞系的转染效率会相差几个数量级(如见 Spandidos and Wilkie 1984)。转染效率决定于细胞类型 [既使同一细胞系的不同克隆或不同传代数也可能出现显著差别(Corsaro and Pearson 1981,Van Pel et al.1985)]、导入 DNA 的性质及相关转录控制信号的功效、用于转染的 DNA 量。   |

更有优质直播、研选好物、福利活动等你来!


我的询价
询价列表

暂时没有已询价产品
手机验证