



Cayman Chemical Company,Inc.
入驻年限:4 年
- 联系人:
Kristina Whitfield
- 所在地区:
美国
- 业务范围:
试剂、抗体、细胞库 / 细胞培养、ELISA 试剂盒
- 经营模式:
生产厂商
推荐产品





公司新闻/正文
TNF-α-Targeted Therapies in Alzheimer’s Disease
人阅读 发布时间:2024-01-29 13:35
Article from 2024-01-17
It is estimated that 99% of drugs in clinical trials for the treatment of Alzheimer's disease fail.1 One of the reasons may be that Alzheimer's disease is a multi-faceted and highly heterogeneous disease, with varying etiological and pathological factors at play.2 Therapies that explore the connection between neuroinflammation and Alzheimer's disease may be a promising new approach for novel treatment strategies.
Introduction
Alzheimer's disease is a neurodegenerative disorder characterized by progressive memory loss, cognitive impairment, behavioral changes, and loss of functional abilities. Alzheimer's disease is the leading cause of dementia, and it is estimated to affect 17% of people aged 75-84 years in the United States.3 Given the growing aging population, it is expected that the incidence will increase in coming years.4 Current treatments for Alzheimer's disease (Table 1) only temporarily alleviate the symptoms of cognitive decline but do not halt the progression of the disease.3,5 Accordingly, there is an urgent need to identify new Alzheimer's disease treatment strategies.
| Compound | Target |
| Donepezil | An AChE inhibitor |
| Tacrine (hydrochloride) | An AChE and butyrylcholinesterase inhibitor |
| Galantamine | An AChE inhibitor |
| Memantine (hydrochloride) | An NMDA receptor antagonist |
| Rivastigmine (tartrate) | A cholinesterase inhibitor |
Table 1. Current therapies for the treatment of Alzheimer's disease.
Pathological Hallmarks of Alzheimer's Disease
Alzheimer's disease is characterized by the formation of amyloid-β (Aβ) plaques and neurofibrillary tangles (NFTs), which are comprised of hyperphosphorylated tau (Figure 1).2,6 These pathological hallmarks are thought to lead to significant neuronal loss and consequent cognitive decline.
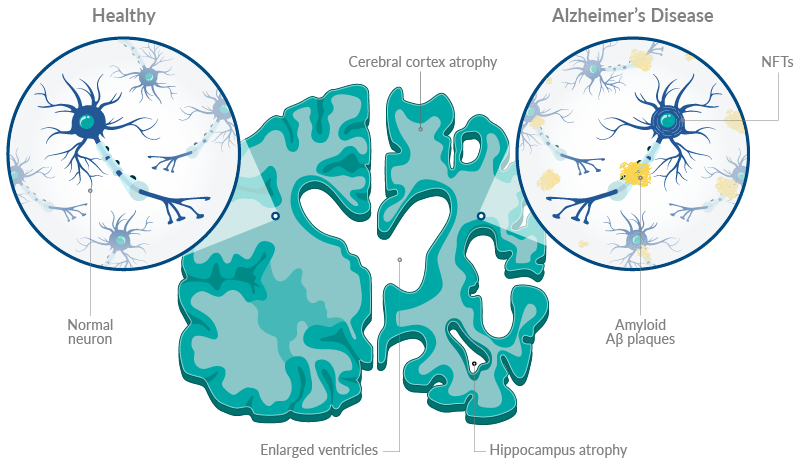
Figure 1. An overview of pathological hallmarks of Alzheimer's disease.
Aβ is a cleavage product of amyloid precursor protein (APP). β-secretase (BACE) and γ-secretase are two enzymes that cleave APP during amyloidogenic processing.2 This produces amyloid-β (Aβ) peptides of various lengths, including Aβ40 and Aβ42, which are neurotoxic peptides found in amyloid plaques.
Under physiological conditions, tau facilitates microtubule formation in axons and has roles in neuronal plasticity.2,6 However, tau hyperphosphorylation results in tau aggregation, producing NFTs that accumulate in axons and dendrites, resulting in neuronal and synaptic loss.7
Given that Aβ plaques are evident decades before NFT formation and memory decline, Aβ-targeting therapies, including β-secretase and γ-secretase inhibitors, have been major drug targets for potential disease-modifying therapies in Alzheimer's disease.8 However, clinical trials employing Aβ-targeted therapies have been largely disappointing.9 Although these agents reduce amyloid plaque formation, they largely do not appear to improve cognitive function or slow disease progression.10
Roles of Neuroinflammation in Alzheimer's Disease
Neuroinflammation is another feature of Alzheimer's disease that may underlie key events that drive initiation and progression of Alzheimer's disease.3,4,10-12Much like Aβ, neuroinflammation has been observed in the brains of patients with Alzheimer's disease decades before the onset of clinical symptoms.2 Hence, targeting neuroinflammation is of great interest in the pursuit of disease-modifying treatments for Alzheimer's disease.
Neuroinflammation is the inflammatory response that occurs in the central nervous system (CNS) in response to many factors, including infection, trauma, ischemia, damage-associated molecular patterns (DAMPs), and, in the case of Alzheimer's disease, Aβ and NFTs.10-12 It is characterized by the production of cytokines, chemokines, reactive oxygen and nitrogen species, and other secondary messengers that recruit and activate immune cells to damaged tissues.
Microglia Drive Neuroinflammation
Macrophage-like cells called microglia are major drivers of neuroinflammation.3,8,10,12 Like macrophages, microglia patrol their environment and react with an appropriate immune response upon sensing a CNS insult. Accordingly, they express several cell receptors for recognizing inflammatory insults and perform key immune functions like phagocytosis and tissue repair.
A growing body of literature suggests that the contribution of microglia to Alzheimer's disease is phase dependent.3,10 Microglial activation early in Alzheimer's disease pathogenesis appears beneficial, as activated microglia eliminate Aβ via phagocytosis and proteolysis, limiting its accumulation, and secrete a wide range of inflammatory mediators that recruit and activate other immune cells, collectively promoting the resolution of inflammation.
However, sustained activation of microglia creates a vicious auto-amplifying cycle that exacerbates neuroinflammation and Alzheimer's disease progression (Figure 2).3,10 The relationship between unresolved inflammation and Aβ accumulation drive this cycle. A major player in neuroinflammation in Alzheimer's disease is TNF-α. Microglia secrete TNF-α in response to stimulation with Aβ, which in turn both reduces microglia phagocytosis of Aβ and upregulates the production of secretases, further driving amyloidogenesis. These TNF-α-driven processes collectively promote Aβ accumulation, which damages neurons and activates microglia. Furthermore, damaged neurons themselves release a host of inflammatory mediators that perpetuate inflammation.
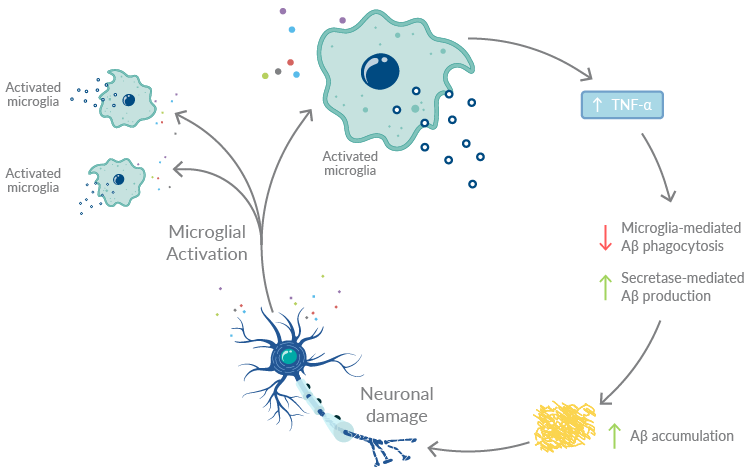
Figure 2. Unresolved neuroinflammation unleashes a vicious cycle that exacerbates inflammation and Alzheimer's disease progression.
TNF-α in Neuroinflammation
TNF-α is a pleiotropic cytokine that has major roles in neurodegenerative diseases, including Alzheimer's disease.3,13,14 It mediates many cellular effects, ranging from cytokine production to roles in cell survival and differentiation, through interactions with its receptors TNFR1 and TNFR2 and downstream signaling cascades, including NF-κB, among others.
Importantly, a large body of evidence indicates that while TNFR1 signaling promotes neuroinflammation (e.g., induces apoptosis and inflammatory responses), TNFR2 signaling is neuroprotective (e.g., promotes cell survival and resolution of inflammation) (Figure 3).13,15-18 TNFR1 is upregulated and TNFR2 is downregulated in postmortem brains from patients with Alzheimer's disease, further suggesting the notion of a link in aberrant TNF-α signaling and Alzheimer's disease.15 Taken together, therapies that selectively inhibit TNFR1 and spare and/or activate TNFR2 could be beneficial.
Indeed, Ortí-Casañ et al. demonstrated that treatment with an anti-TNFR1 antibody reduces neuronal loss and memory deficits in a mouse model of neurodegeneration.16 The same group also showed that a TNFR2 agonist improves cognitive function and reduces Aβ plaque load in a transgenic mouse model of Alzheimer's disease.18
Another approach to selectively target TNFR1 is through inhibition of a disintegrin and metalloprotease domain 17 (ADAM17), also known as TNF-α-converting enzyme (TACE). TNF-α is synthesized as a membrane-bound protein (mTNF-α) that is cleaved by ADAM17 to produce soluble TNF-α (sTNF-α).19 Intriguingly, while TNFR1 is activated by both sTNF-α and mTNF-α, TNFR2 is preferentially activated by mTNF-α. Hence, ADAM17 inhibition reduces sTNF-α production, limiting consequent TNFR1 activation while sparing TNFR2 activation.
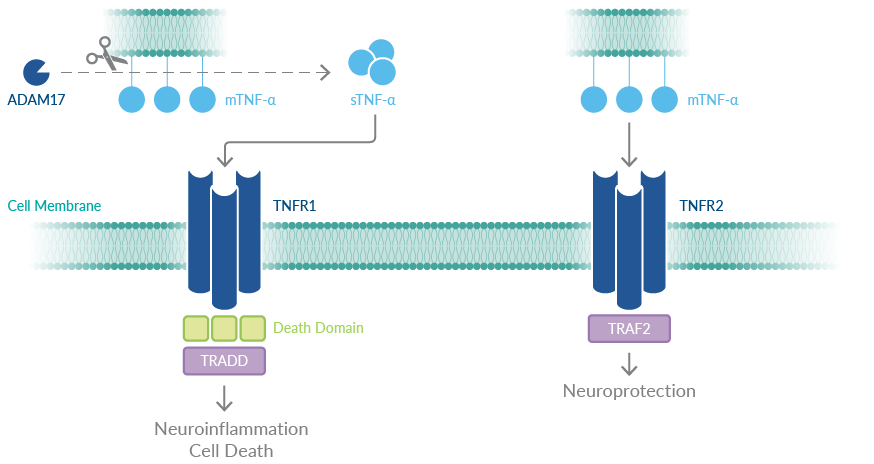
Figure 3. Simplified summary of TNFR signaling roles in the brain.
Current Therapies Targeting TNF-α in Alzheimer's Disease
Because of TNF-α's critical role as a central mediator of inflammation, agents that block TNF-α are attractive therapies for a wide variety of inflammatory conditions.
Anti-TNF-α therapies are used clinically in the treatment of rheumatoid arthritis and psoriasis.19 In retrospective epidemiological studies, patients with rheumatoid arthritis or psoriasis treated with TNF-α-blocking therapies for these conditions had reduced risk of Alzheimer's disease, suggesting that TNF-α blockade is a possible avenue for preventative and/or disease-modifying therapies for Alzheimer's disease.14,20
While epidemiological studies and preclinical models of Alzheimer's disease generally support the notion that anti-TNF-α-based therapies are beneficial in Alzheimer's disease, a double-blind, phase 2 clinical trial employing the TNF-α-blocking therapy etanercept did not find any improvement in cognitive function.21Given the heterogeneity of Alzheimer's disease and the multiple functions of TNF-α and its receptors, future studies that consider the full spectrum of TNF-α-driven activities could lead to more promising results.
Necroptosis Drives Neuronal Loss in Alzheimer's Disease
A newly discovered mechanism of neuronal cell death in Alzheimer's disease could prove to be another TNF-α-related approach for new Alzheimer's disease therapies. For many years, how neurons die in Alzheimer's disease was unknown. A groundbreaking study by Balusu and colleagues revealed that neuronal cell death in a human neuron xenograft mouse model of Alzheimer's disease was driven by necroptosis, an inflammatory form of cell death.22 In this study, inhibition of necroptosis with ponatinib or dabrafenib protected against neuronal loss in this preclinical model. As such, inhibition of necroptosis may also represent a new therapeutic strategy for Alzheimer's disease.
TNF-α Induces Necroptosis
Necroptosis is a form of programmed cell death with features resembling both apoptosis and necrosis. It results in cell swelling and eventual cell lysis, culminating in the release of a milieu of DAMPs that further promote neuroinflammation.23 Necroptosis has been observed in postmortem brains from patients with Alzheimer's disease, where it positively correlates with disease stage and inversely correlates with cognitive function.24
Necroptosis is initiated by several inflammatory ligands that activate death receptors expressed on the surface of cell membranes.23 TNF-α is the best characterized inflammatory trigger of necroptosis. As TNFR2 does not contain a death domain (see Figure 3 above), most research has focused on the role of TNFR1 in necroptosis.25
In brief, upon stimulation with TNF-α, TNFR1 forms a necrosome, which consists of receptor-interacting protein kinase 1 (RIPK1), RIPK3, and mixed lineage kinase domain-like protein (MLKL).26 RIPK1 initiates a phosphorylation cascade that leads to phosphorylation of MLKL, which translocates to the plasma membrane and forms a pore, resulting in cell rupture and subsequent release of cytokines and other inflammatory mediators. Importantly, RIPK1 does not signal through the TNFR2-mediated pathways, indicating that therapies inhibiting this kinase spare TNFR2 signaling and preserve the critical neuroprotective effects of TNFR2 signaling.27
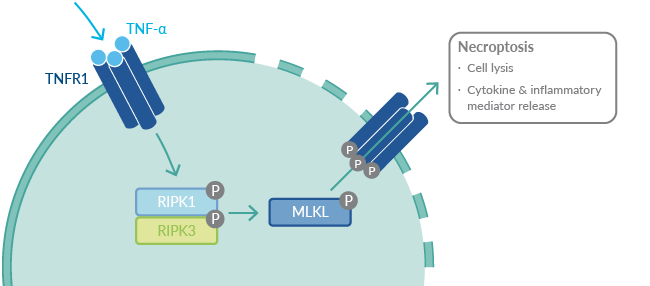
Figure 4. A simplified view of signaling events leading to necroptosis.
Accordingly, RIPK1 inhibitors are one avenue being explored for new Alzheimer's disease therapies. A clinical trial employing a RIPK1 inhibitor for the treatment of neurodegenerative disorders is currently underway. SAR443820, formerly known as DNL788, is a potent and brain-penetrant inhibitor of RIPK1 with a favorable safety profile that is currently in a phase 2 clinical trial in patients with amyotrophic lateral sclerosis (ALS).28 It is anticipated that SAR443820, should it perform well in this patient cohort, will also have benefits translatable to other neurodegenerative disorders, including Alzheimer's disease.
Future Directions
Delineating the precise contributions of TNF-α in the onset and progression of Alzheimer's disease has been challenging given the heterogeneity of Alzheimer's disease and the multiple roles of TNF-α, which can have protective or harmful effects in a context-dependent manner.
Cayman offers a wide range of products for the elucidation of neuroinflammation in Alzheimer's disease, including contributions through TNF-α signaling pathways and necroptosis.
询价列表

暂时没有已询价产品
