


推荐产品





公司新闻/正文
你有一份 HRD 分析指南待查收!
人阅读 发布时间:2021-08-05 17:12
背景
对人类肿瘤组织样本的同源重组缺陷(Homologous Recombination Deficiency, HRD)进行可靠量化具有重要的临床意义。伴有 BRCA1 或 BRCA2 缺失的卵巢癌和三阴性乳腺癌对 PARP 抑制剂和铂类化疗高度敏感,并以 DNA 总拷贝数畸变的形式显示基因组瘢痕的积聚;没有 BRCA1 或 BRCA2 丢失,但有类似基因组疤痕积聚的癌种也显示出对铂类化疗的敏感性增加[1-5]。此前基于 SNP 阵列的染色体不稳定性特征:杂合性丢失(loss of heterozygosity,LOH)、端粒等位基因失平衡(telomeric allelic imbalance,TAI)和大片段迁移(large-scale state transitions,LST),其综合得分可作 HRD 的生物标志物。近来二代测序法,如全基因组测序(Whole Genome Sequencing, WGS)和全外显子测序(Whole Exome Sequencing, WES)逐渐取代 SNP 芯片成为基因组瘢痕分析的主流方法[6]。
考虑到肿瘤标本的倍性、纯度和异质性,为了满足高覆盖率和低成本的要求,基于二代测序的靶向测序(Targeted Next-Generation Sequencing, Tg-NGS)锚向全基因组内均匀分布、高杂合率的数万个 SNP 位点亦可以应用于同源重组缺陷的检测分析。然而,WES 和 Tg-NGS 数据捕获范围相差较大,分析方法也不尽相同。纳昂达此前推出的 HiSNP Ultra Panel 是一款基于 SNP 且全基因组范围内 50 Kb 均匀分布的 Panel,可应用于 HRD 检测分析。借助 HRD 标准品,本文评估了纳昂达科技 Panel 在 HRD 评估中的表现,以期为用户提供参考。
材料与方法
HRD 标准品及其参考值,来源于菁良(菁良基因科技(深圳)有限公司),每对标准品均含有肿瘤样本及其对照样本,具体信息见表1。每个 DNA 样本取 50 ng 并片段化至 ~250 bp,分别使用 NadPrep® for Illumina/MGI 系列文库构建试剂盒,构建预文库。预文库按照纳昂达标准杂交流程分别进行 WES(Exome Plus Panel v2.0,Exome+)和 Tg-NGS(HiSNP Ultra Panel v1.0,HiSNP Ultra)的捕获。杂交后的文库分别在 Illumina 或者 MGISEQ 平台测序,WES 测序深度 ~300x,Tg-NGS 测序深度 ~400x。WGS 数据平均测序深度为 40x(菁良提供)。
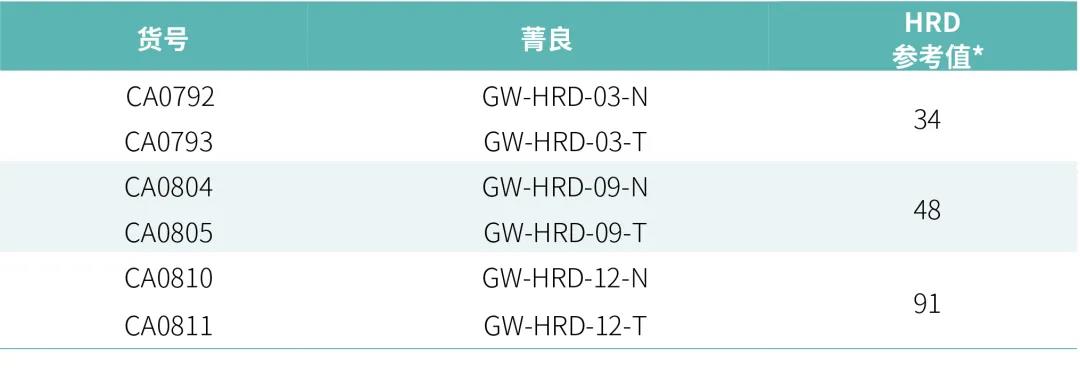
表1:HRD 标准品信息
ASCAT[7], sequenza[8] 和 facets[9] 用于捕获测序数据的等位基因特异拷贝数(Allele Specific Copy Number, ASCN)分析;TITAN[10] 用于WGS 数据的 ASCN 分析;NAD-HRD(纳昂达内部分析软件)和 scarHRD 用于拷贝数变异基础上的 HRD 分析。所有软件分析均使用默认参数,软件信息见表2。
表 2:HRD 分析方式及软件

WGS 和 Tg-NGS 应用于 HRD 标准品的评估
HRD 标准品的 Tg-NGS 数据显示平均中靶率均>88%,平均深度>300x。部分标准品中肿瘤样本的 0.2x 平均覆盖深度区域略低于对照样本(~96% vs ~98%),提示其肿瘤异质性高。
我们首先使用 ASCAT、sequenza、facets 和 TITAN 软件对标准品进行了纯度和倍性分析。值得注意的是,这四款软件分析均要求为配对样本。不同 Panel 计算结果显示,纯度基本一致,即肿瘤标准品的纯度均接近 1。而在倍性分析中,WGS、WES 和 Tg-NGS 的不同软件计算结果有所差异。Tg-NGS 捕获在不同样本分析时也存在较大差异,推测与不同软件分析的算法有关。ASCAT 在计算偏高 HRD 值的标准品倍性时与其他软件存在明显差异。
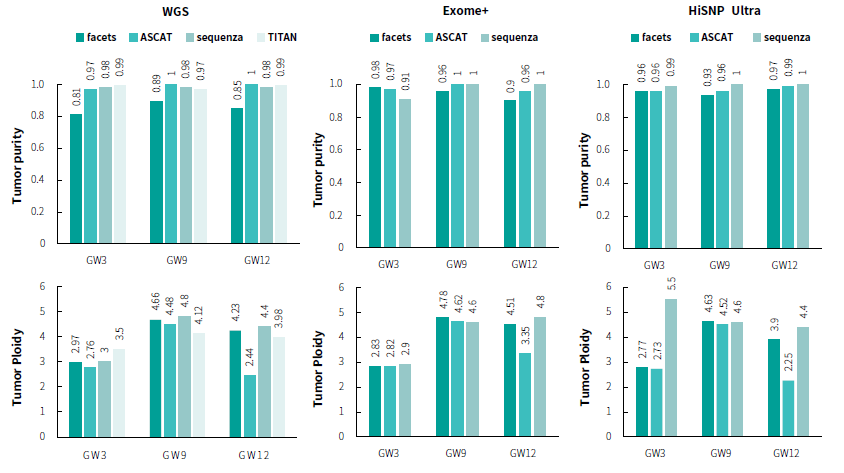
图 1:菁良标准品的纯度和倍性分析
当计算得出 HRD 标准品的纯度和倍性后,进行 ASCN 分析流程。纳昂达根据公开文献中 LOH、LST 和 TAI 的算法,开发了搭配 ASCAT 使用分析HRD score 软件:NAD-HRD。而facets、sequenza 和 TITAN 则使用 scarHRD 分析 HRD Score。从 HRD 分析结果来看,Tg-NGS 和 WGS 的 HRD 检测结果趋势判断与标准品参考值相一致(图2)。Tg-NGS 的 HRD 分析中,采用 sequenza (scarHRD 内置)分析 ASCN 值进而判断 HRD Score 的方法较符合参考值。而 WGS 的 HRD 分析采用经典 WGS CNV 检测软件 TITAN,结果比 Tg-NGS 更接近参考值。
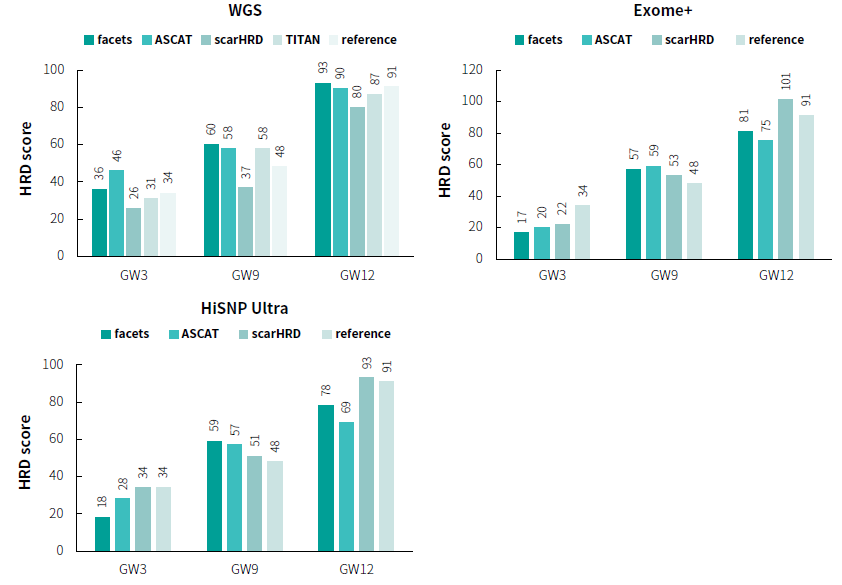
图 2:菁良标准品 HRD 分析
分辨率对 HRD 标准品评估的影响
测序分析区域的不同分辨率会产生 ASCN 及 HRD 的结果差异。为了进一步分析 Tg-NGS 和 WGS 在检测 CNV 和 LOH 上的差异,我们选取菁良 HRD 标准品,评估使用 scarHRD 及内置 sequenza 软件检测 ASCN 时的异同。Tg-NGS 的 ASCN 跟 WGS 的分布有着明显差异(图 3 A-C)。WGS分析结果中有更多的短小区域,其主要原因可能是 WGS 数据覆盖全基因组且覆盖均一,在局部区域的分辨率比 Tg-NGS 高。因此,我们采取合并 WGS 内 ASCN segment 间隔小于 1M 的区域,再与 Tg-NGS 比较检测区域的重现性(图 3 D)。Tg-NGS 在Low-HRD 的样本中 ASCN 区域的重现性较好(≥94%,GW3),而对于 High HRD 的样本则效果略差,说明复杂的基因组变化采用非全基因组覆盖的方式仍会有一定的误差。
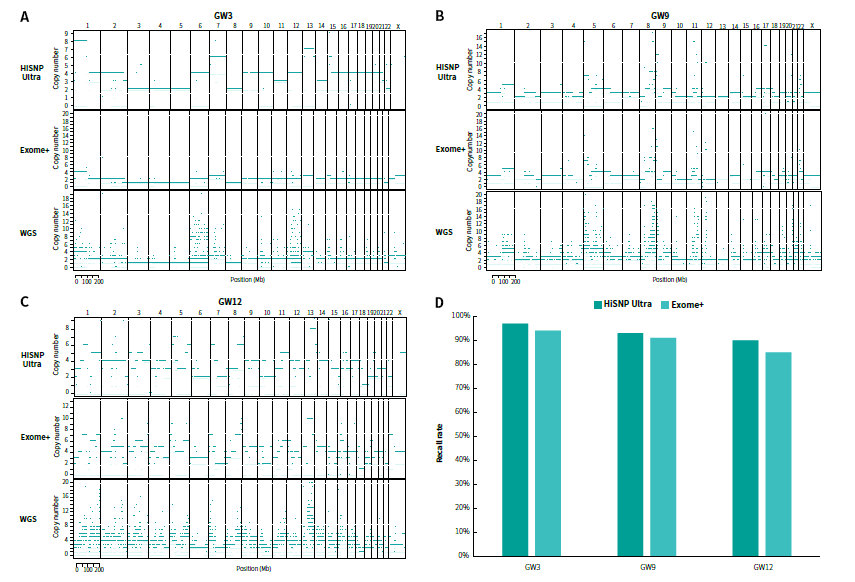
图 3:菁良 HRD 标准品 ASCN 分析结果
Tg-NGS 方法中,HiSNP Ultra 和 Exome+ 的 ASCN 在全基因组范围上较为接近;然而,HiSNP Ultra 对 WGS 的重现性均优于 Exome+。我们将全基因组划分为数个 3Mb 区域,并统计不同标准品在单个区域内的杂合位点个数,筛选条件为:>0.2x 平均覆盖深度且 VAF=0.3-0.7。如图 4 所示,HiSNP Ultra 捕获的杂合位点数量集中在 22-24 个/3 Mb,而 Exome+ 捕获的杂合位点数量则集中在 0-4 个/3 Mb(图 4)。这表明 HiSNP Ultra 能检测更多、更均匀的杂合位点用于检测 ASCN 和 HRD。
另外,由于 HRD 的肿瘤标准品中存在多个 LOH,因此肿瘤标准品在 3 Mb 区域内检测的杂合位点数要小于对照样本。这意味着 HiSNP Ultra 比 Exome+ 能更加明显的反映肿瘤和对照样本之间杂合位点的差异,提高 ASCN 和 LOH 的检测效率。
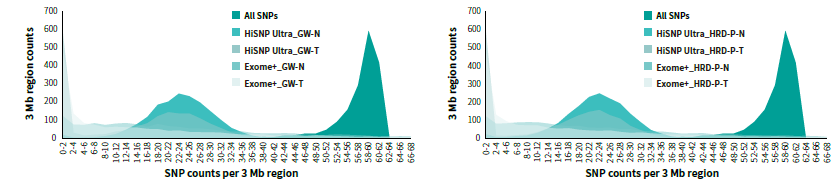
图 4:HRD 标准品 3M 区域平均杂合位点数分布
不同肿瘤纯度对 HRD 标准品评估的影响
真实世界的肿瘤组织样本中均含有一定的正常细胞,样本纯度对肿瘤倍性、拷贝数变异的分析至关重要。面对纯度不高的样本,HRD 检测 Panel 和分析工具的检测能力受到挑战。为验证 HRD 分析与纯度阈值的关系,我们进行了纯度模拟分析(in silicon) :将 HRD 标准品的肿瘤样本数据和对照样本数据按照一定比例(10%-90%)混合后,再进行纯度、倍性和 HRD 值的计算。结果显示纯度的计算结果与理论值呈线性关系。从标准品模拟数据可见,肿瘤样本在肿瘤纯度达到 30% 以上时其倍性值和 HRD 值趋于稳定。我们推荐 HiSNP Ultra 的纯度阈值在 30% 或以上。

图 5:菁良 HRD 标准品 in silicon 纯度模拟分析
不同测序深度对 HRD 标准品评估的影响
等位基因特异性拷贝数的稳定评估和测序深度有关,因此sequenza、facets 和 TITAN 等软件对 CNV 和 ASCN 的计算对测序深度都有一定要求。我们对标准品的测序数据随机取样(downsampling),以模拟不同测序深度的子集(50x,100x, 150x, 200x, 250x)时,对 HRD 分析的影响。在 100%肿瘤纯度的 HRD 标准品中,≥50x 深度时即可准确进行纯度和倍性的分析,此时 HRD 分析结果也基本与参考值相一致(图6)。
考虑到真实世界中的样本情形,我们同时模拟了 30% 和 40% 纯度梯度测序数据,以评估低纯度下肿瘤样本测序深度对 HRD 分析的影响。由于纯度的计算依赖于 BAF(B-Allele Frequency),较低深度会导致某些位点的 BAF 计算不够准确,从而降低纯度结果的准确性。当测序深度 ≥200x,纯度数据才趋于稳定。另外,HRD 本身也会导致某些位点信息的丢失,使得纯度计算结果均低于理论值。不同测序深度对于倍性分析影响的较小,≥100x 时结果非常稳定。
HRD 分析结果显示不同深度下 HRD 值存在较大差异,当测序深度 ≥200x 时 HRD 才趋于稳定。与之同时,肿瘤本身 HRD 值的高低也会影响差异的大小:Low-HRD 的样本在不同深度时的计算结果相差较小;High-HRD 的样本相差很大,可能影响到对 HRD 类别判定。因此,HiSNP Ultra 面对纯度不一的真实样本时,应保证足够的测序深度,方能稳定分析 HRD。我们推荐原始测序原始深度不低于300x。
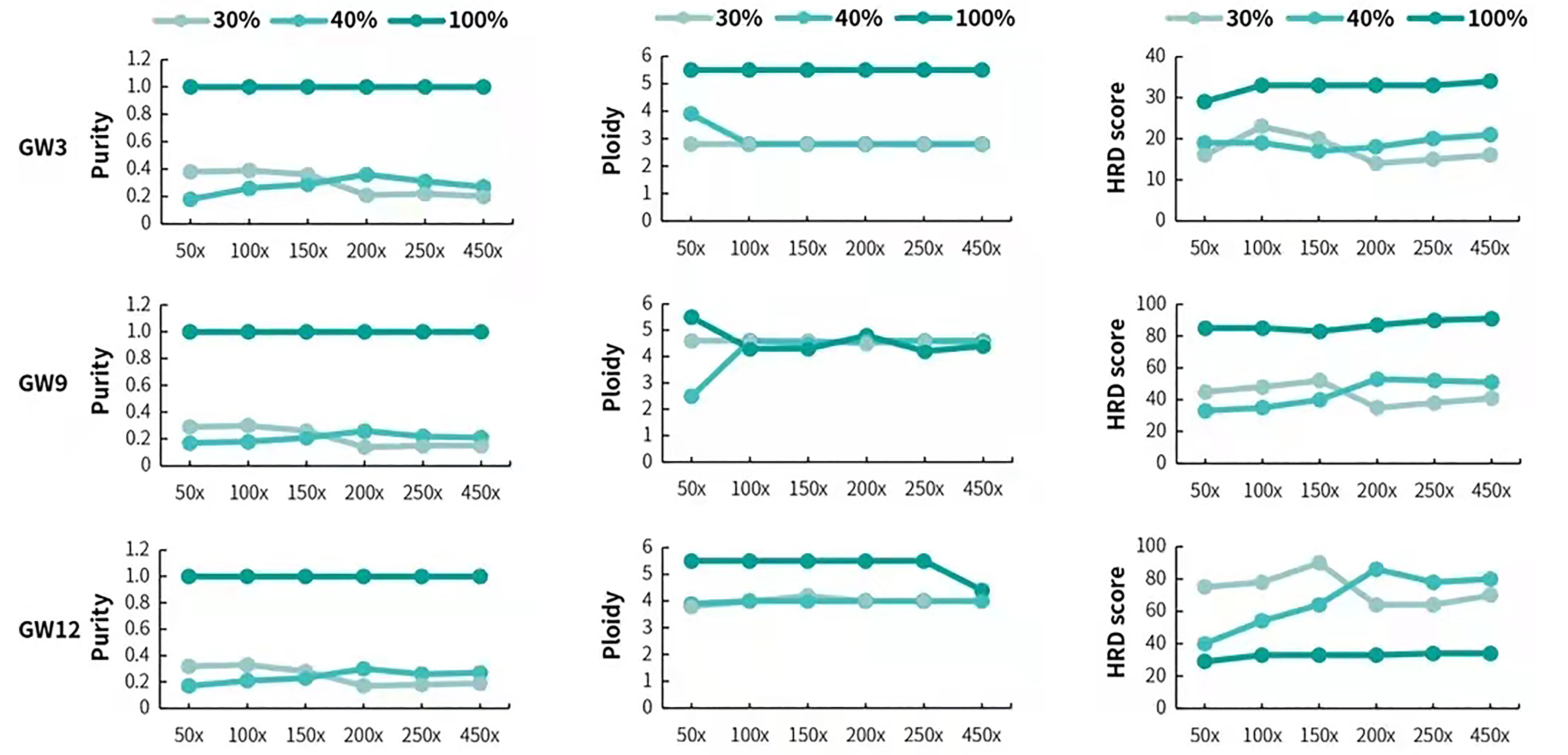
图 6:菁良 HRD 标准品不同纯度梯度下低深度子集 HRD 计算情况
总结
早前实验表明基于 SNP 和 WGS/WES 的 HRD 值相关性良好。其中,TAI、LST 和 HRD 总值无显著差异,但基于 WGS/WES 的 LOH 事件数显著减少,可能是由于片段分割算法的不同或样本质量、覆盖率较低[6]。纳昂达推出的 HiSNP Ultra Panel 是一款基于内含子 SNP 且全基因组范围内 50 Kb 均匀分布的基因检测组套,可应用于 HRD 检测分析。在本文中,我们基于此 Panel 进行了 HRD 标准品评估对比分析,并发现可达到与 WGS 相一致的 HRD 分析结果,并比 WES 更优。
HRD 评分在不同起源器官和组织类型之间差异很大[11]。此外,不同分析软件对 LOH、LST 和 TAI 等计算方法和阈值选择并不相同,分析的 HRD 值亦可能存在差异。但整体上多款软件的 HRD 分析结果的趋势基本一致,其中 scarHRD 软件的计算结果更为接近标准品的参考值。
ASCN 分析是 HRD 值计算的基础,与肿瘤样本纯度和测序深度密切相关。我们发现 HiSNP Ultra 在肿瘤纯度 ≥30%,测序深度 ≥200x 时,方能稳定的用于 HRD 检测。
面向真实的 HRD 评估的应用场景,对含 BRCA 在内的同源重组修复通路(homologous recombination repair, HRR)基因的变异检测是必要的。纳昂达科技的 HRR Panel v1.0 靶向 HRR 相关的 35个 HRR 基因的编码区,可 spike-in 至 HiSNP Ultra,实现基于基因突变检测和基因组不稳定状态打分同时进行的完整 HRD 评估方案。
本文节选自《纳昂达 Panel HRD 分析指南》,限于篇幅未展示南京科佰的标准品结果。如您需要全文,请留言或联系 support@njnad.com。
相关试剂信息

参考文献
[1] Telli M L, Timms K M, Reid J, et al. Homologous recombination deficiency(HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer[J]. Clinical cancer research, 2016, 22(15): 3764-3773.
[2] Mirza M R, Monk B J, Herrstedt J, et al. Niraparib maintenance therapyin platinum-sensitive, recurrent ovarian cancer[J]. New England Journal of Medicine, 2016, 375(22): 2154-2164.
[3] Marquard A M, Eklund A C, Joshi T, et al. Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs[J]. Biomarker research, 2015, 3(1): 1-10.
[4] den Brok W D, Schrader K A, Sun S, et al. Homologous recombination deficiency in breast cancer: a clinical review[J]. JCO Precision Oncology, 2017, 1: 1-13.
[5] Lotan T L, Kaur H B, Salles D C, et al. Homologous recombination deficiency (HRD) score in germline BRCA2-versus ATM-altered prostate cancer[J]. Modern Pathology, 2021: 1-9.
[6] Sztupinszki Z, Diossy M, Krzystanek M, et al. Migrating the SNP array-based homologous recombination deficiency measures to next generation sequencing data of breast cancer[J]. NPJ breast cancer, 2018, 4(1): 1-4.
[7] Ross E M, Haase K, Van Loo P, et al. Allele-specific multi-sample copy number segmentation in ASCAT[J]. Bioinformatics, 2020.
[8] Favero F, Joshi T, Marquard A M, et al. Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data[J]. Annals of Oncology, 2015, 26(1): 64-70.
[9] Shen R, Seshan V E. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing[J]. Nucleic acids research, 2016, 44(16): e131-e131.
[10] Ha G, Roth A, Khattra J, et al. TITAN: inference of copy number architectures in clonal cell populations from tumor whole-genome sequence data[J]. Genome research, 2014, 24(11): 1881-1893.
[11] Takaya H, Nakai H, Takamatsu S, et al. Homologous recombination deficiency status-based classification of high-grade serous ovarian carcinoma[J]. Scientific reports, 2020, 10(1): 1-8.
[1] Telli M L, Timms K M, Reid J, et al. Homologous recombination deficiency(HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer[J]. Clinical cancer research, 2016, 22(15): 3764-3773.
[2] Mirza M R, Monk B J, Herrstedt J, et al. Niraparib maintenance therapyin platinum-sensitive, recurrent ovarian cancer[J]. New England Journal of Medicine, 2016, 375(22): 2154-2164.
[3] Marquard A M, Eklund A C, Joshi T, et al. Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs[J]. Biomarker research, 2015, 3(1): 1-10.
[4] den Brok W D, Schrader K A, Sun S, et al. Homologous recombination deficiency in breast cancer: a clinical review[J]. JCO Precision Oncology, 2017, 1: 1-13.
[5] Lotan T L, Kaur H B, Salles D C, et al. Homologous recombination deficiency (HRD) score in germline BRCA2-versus ATM-altered prostate cancer[J]. Modern Pathology, 2021: 1-9.
[6] Sztupinszki Z, Diossy M, Krzystanek M, et al. Migrating the SNP array-based homologous recombination deficiency measures to next generation sequencing data of breast cancer[J]. NPJ breast cancer, 2018, 4(1): 1-4.
[7] Ross E M, Haase K, Van Loo P, et al. Allele-specific multi-sample copy number segmentation in ASCAT[J]. Bioinformatics, 2020.
[8] Favero F, Joshi T, Marquard A M, et al. Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data[J]. Annals of Oncology, 2015, 26(1): 64-70.
[9] Shen R, Seshan V E. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing[J]. Nucleic acids research, 2016, 44(16): e131-e131.
[10] Ha G, Roth A, Khattra J, et al. TITAN: inference of copy number architectures in clonal cell populations from tumor whole-genome sequence data[J]. Genome research, 2014, 24(11): 1881-1893.
[11] Takaya H, Nakai H, Takamatsu S, et al. Homologous recombination deficiency status-based classification of high-grade serous ovarian carcinoma[J]. Scientific reports, 2020, 10(1): 1-8.
询价列表

暂时没有已询价产品

快捷询价 发送名片
当你希望让更多商家联系你时,可以勾选后发送询价,平台会将你的询价消息推荐给更多商家。