- 移动端


上海诺佰佰生物科技有限公司
4 年
手机商铺
- NaN
- 0.5
- 0.5
- 2.5
- 2.5
推荐产品

技术资料/正文
N家目录 | HCD检测试剂盒定制开发服务
174 人阅读发布时间:2025-06-12 11:03
1.HCD介绍
生物制品在生产的过程中,其宿主细胞DNA的残留,可能会造成插入突变,导致抑癌基因失活、癌基因被激活等危害。这会严重影响生物制品的安全性。各国药品监督管理机构对宿主细胞DNA的残留量有着严格的限度控制,同时各国药典也提供数种经典的检测方法,建立合适的宿主细胞残留DNA检测方法有助于监测生产工艺, 确保生物制品的安全性。
2.HCD检测不同试剂盒类型(政策法规/方法平台)
各国法规药典对残留检测的要求:
|
分类 |
要求项目 |
中国标准 |
国际标准 |
检测方法 |
其他说明 |
|
残留量限制要求 |
细胞基质(CHO、Vero) |
≤100 pg/剂 |
FDA/WHO:≤100 pg/剂;大剂量生物制品(如单抗)可放宽至10 ng/剂;欧盟标准与中国类似,特定疫苗更严 |
- |
基因治疗产品部分企业内控标准更严(≤0.1 ng/剂) |
|
细菌/真菌基质 |
≤10 ng/剂 |
||||
|
片段大小控制要求 |
片段长度限制 |
HCD片段≤200bp(NMPA要求) |
FDA:HCD片段≤200bp |
毛细管凝胶电泳-激光诱导荧光法(CGE-LIF),检测范围50-1000bp |
片段越小安全性越高(功能基因需≥200bp才可能致病) |
|
检测方法法规要求 |
中国药典认可方法 |
qPCR(灵敏度1 fg/μL,符合药典三部通则3407) |
- |
DNA探针杂交法、荧光染色法(灵敏度低,逐步淘汰) |
方法学验证需符合ICH Q2(R2)(准确性、精密度、线性范围) |
|
试剂盒验证要求 |
灵敏度(LLOD)、特异性(无交叉反应)、回收率90-100% |
||||
|
国际协调与双报要求 |
法规协调 |
残留量标准与FDA/WHO一致 |
中美欧残留量要求基本一致;片段大小要求可能需调整 |
推荐使用符合多国药典的检测服务(如恒驭生物HCD检测服务) |
生产工艺需结合深层过滤、层析技术优化参数,检测数据需覆盖全流程(研发至生产) |
各国法规对HCD残留量均有严格限制,但商业化的通用HCD检测试剂盒通常难以覆盖所有场景,需要考虑结合宿主细胞类型及工艺特异性等因素,在复杂生物制品中推荐使用平台化方法或针对上游工艺开发的产品/工艺专属性定制化开发的产品能更好的满足研发需求。
3.NovoBio提供定制化服务
NovoBio 基于探针法的荧光定量PCR平台,可提供HCD试剂盒开发和验证服务。
- 开发流程

- 服务周期
总服务时长:2.5 M-3.5 M

- 应用案例
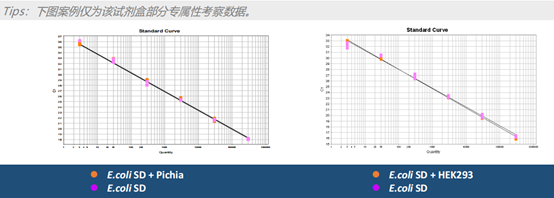
(1)专属性验证:
针对NovoBio【KT6008 E.coli 残留DNA定量试剂盒】,对其标准品中加入Pichia、HEK293的DNA样本,考察非目的片段的加入对标准曲线的影响。
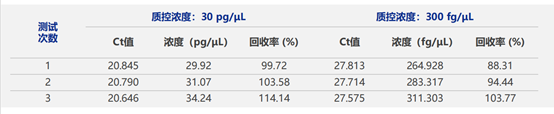
(2)准确度验证:
在线性范围内选用NovoBio【KT6008 E.coli 残留DNA定量试剂盒】检测30 pg/μL、300 fg/μL E.coli 国家标准品DNA,重复测定3次。可接受标准:检测浓度值测量偏差绝对值<10%。
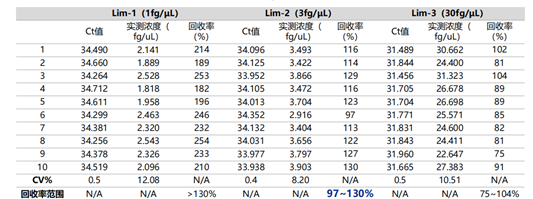
(3)定量限验证:
选用NovoBio【KT6008 E.coli 残留DNA定量试剂盒】重复检测1 fg/μL、3 fg/μL、30 fg/μL E.coli DNA各10次,满足CV%<20%且回收率在70%-130%的浓度,即为定量限。
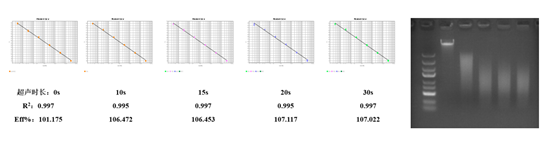
(4)耐用性验证:
选用NovoBio【KT6008 E.coli 残留DNA定量试剂盒】进行检测。取试剂盒中E.coli标准品5份,1份作为对照, 4份分别进行 10s、15s、20s、30s不同时长的超声处理,将这些DNA 分别进行稀释,进行线性测试。
- 平台优势
✦成熟的HCD产品开发平台:已成功开发数十种检测不同物种的HCD产品
✦优质的产品质量:灵敏度达fg级别,参照中国药典生物制品标准品标定方法
✦无忧的售前售后保障:售前可提供免费样本测试,售后有专业的技术支持团队
✦一站式服务:可提供符合FDA GLP 法规的验证服务
- 相关产品
|
应用场景 |
货号 |
产品名称 |
规格 |
|
宿主细胞残留DNA检测 |
KT6009 |
CHO残留DNA定量试剂盒 |
96 T/盒 |
|
KT6008 |
E.coli残留DNA定量试剂盒 |
96 T/盒 |
|
|
KT6024 |
毕赤酵母残留DNA定量试剂盒 |
96 T/盒 |